Here you’ll find a short description and examples of how to use the FASTX-toolkit from the command line.
Command Line Arguments
Most tools show usage information with -h.
Tools can read from STDIN and write to STDOUT, or
from a specific input file (-i) and specific output file (-o).
Tools can operate silently (producing no output if everything was OK), or
print a short summary (-v).
If output goes to STDOUT, the summary will be printed to STDERR.
If output goes to a file, the summary will be printed to STDOUT.
Some tools can compress the output with GZIP (-z).
FASTQ-to-FASTA
1 | $ fastq_to_fasta -h |
FASTX Statistics
$ fastx_quality_stats -h
usage: fastx_quality_stats [-h] [-i INFILE] [-o OUTFILE]
version 0.0.6 (C) 2008 by Assaf Gordon (gordon@cshl.edu)
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
If FASTA file is given, only nucleotides
distribution is calculated (there's no quality info).
[-o OUTFILE] = TEXT output file. default is STDOUT.
The output TEXT file will have the following fields (one row per column):
column = column number (1 to 36 for a 36-cycles read solexa file)
count = number of bases found in this column.
min = Lowest quality score value found in this column.
max = Highest quality score value found in this column.
sum = Sum of quality score values for this column.
mean = Mean quality score value for this column.
Q1 = 1st quartile quality score.
med = Median quality score.
Q3 = 3rd quartile quality score.
IQR = Inter-Quartile range (Q3-Q1).
lW = 'Left-Whisker' value (for boxplotting).
rW = 'Right-Whisker' value (for boxplotting).
A_Count = Count of 'A' nucleotides found in this column.
C_Count = Count of 'C' nucleotides found in this column.
G_Count = Count of 'G' nucleotides found in this column.
T_Count = Count of 'T' nucleotides found in this column.
N_Count = Count of 'N' nucleotides found in this column.
max-count = max. number of bases (in all cycles)
FASTQ Quality Chart
$ fastq_quality_boxplot_graph.sh -h
Solexa-Quality BoxPlot plotter
Generates a solexa quality score box-plot graph
Usage: /usr/local/bin/fastq_quality_boxplot_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "solexa_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title (usually the solexa file name) - will be plotted on the graph.
FASTA/Q Nucleotide Distribution
$ fastx_nucleotide_distribution_graph.sh -h
FASTA/Q Nucleotide Distribution Plotter
Usage: /usr/local/bin/fastx_nucleotide_distribution_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "fastx_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title - will be plotted on the graph.
FASTA/Q Clipper
$ fastx_clipper -h
usage: fastx_clipper [-h] [-a ADAPTER] [-D] [-l N] [-n] [-d N] [-c] [-C] [-o] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-a ADAPTER] = ADAPTER string. default is CCTTAAGG (dummy adapter).
[-l N] = discard sequences shorter than N nucleotides. default is 5.
[-d N] = Keep the adapter and N bases after it.
(using '-d 0' is the same as not using '-d' at all. which is the default).
[-c] = Discard non-clipped sequences (i.e. - keep only sequences which contained the adapter).
[-C] = Discard clipped sequences (i.e. - keep only sequences which did not contained the adapter).
[-k] = Report Adapter-Only sequences.
[-n] = keep sequences with unknown (N) nucleotides. default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-D] = DEBUG output.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Renamer
$ fastx_renamer -h
usage: fastx_renamer [-n TYPE] [-h] [-z] [-v] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.10 by A. Gordon (gordon@cshl.edu)
[-n TYPE] = rename type:
SEQ - use the nucleotides sequence as the name.
COUNT - use simply counter as the name.
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Trimmer
$ fastx_trimmer -h
usage: fastx_trimmer [-h] [-f N] [-l N] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-f N] = First base to keep. Default is 1 (=first base).
[-l N] = Last base to keep. Default is entire read.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Collapser
$ fastx_collapser -h
usage: fastx_collapser [-h] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-v] = verbose: print short summary of input/output counts
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTQ/A Artifacts Filter
$ fastx_artifacts_filter -h
usage: fastq_artifacts_filter [-h] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-z] = Compress output with GZIP.
[-v] = Verbose - report number of processed reads.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
FASTQ Quality Filter
$ fastq_quality_filter -h
usage: fastq_quality_filter [-h] [-v] [-q N] [-p N] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-q N] = Minimum quality score to keep.
[-p N] = Minimum percent of bases that must have [-q] quality.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
FASTQ/A Reverse Complement
$ fastx_reverse_complement -h
usage: fastx_reverse_complement [-h] [-r] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA Formatter
$ fasta_formatter -h
usage: fasta_formatter [-h] [-i INFILE] [-o OUTFILE] [-w N] [-t] [-e]
Part of FASTX Toolkit 0.0.7 by gordon@cshl.edu
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-w N] = max. sequence line width for output FASTA file.
When ZERO (the default), sequence lines will NOT be wrapped -
all nucleotides of each sequences will appear on a single
line (good for scripting).
[-t] = Output tabulated format (instead of FASTA format).
Sequence-Identifiers will be on first column,
Nucleotides will appear on second column (as single line).
[-e] = Output empty sequences (default is to discard them).
Empty sequences are ones who have only a sequence identifier,
but not actual nucleotides.
Input Example:
>MY-ID
AAAAAGGGGG
CCCCCTTTTT
AGCTN
Output example with unlimited line width [-w 0]:
>MY-ID
AAAAAGGGGGCCCCCTTTTTAGCTN
Output example with max. line width=7 [-w 7]:
>MY-ID
AAAAAGG
GGGTTTT
TCCCCCA
GCTN
Output example with tabular output [-t]:
MY-ID AAAAAGGGGGCCCCCTTTTAGCTN
example of empty sequence:
(will be discarded unless [-e] is used)
>REGULAR-SEQUENCE-1
AAAGGGTTTCCC
>EMPTY-SEQUENCE
>REGULAR-SEQUENCE-2
AAGTAGTAGTAGTAGT
GTATTTTATAT
FASTA Nucleotides Changer
$ fasta_nucleotide_changer -h
usage: fasta_nucleotide_changer [-h] [-z] [-v] [-i INFILE] [-o OUTFILE] [-r] [-d]
version 0.0.7
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-v] = Verbose mode. Prints a short summary.
with [-o], summary is printed to STDOUT.
Otherwise, summary is printed to STDERR.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-r] = DNA-to-RNA mode - change T's into U's.
[-d] = RNA-to-DNA mode - change U's into T's.
FASTA Clipping Histogram
$ fasta_clipping_histogram.pl
Create a Linker Clipping Information Histogram
usage: fasta_clipping_histogram.pl INPUT_FILE.FA OUTPUT_FILE.PNG
INPUT_FILE.FA = input file (in FASTA format, can be GZIPped)
OUTPUT_FILE.PNG = histogram image
FASTX Barcode Splitter
$ fastx_barcode_splitter.pl
Barcode Splitter, by Assaf Gordon (gordon@cshl.edu), 11sep2008
This program reads FASTA/FASTQ file and splits it into several smaller files,
Based on barcode matching.
FASTA/FASTQ data is read from STDIN (format is auto-detected.)
Output files will be writen to disk.
Summary will be printed to STDOUT.
usage: /usr/local/bin/fastx_barcode_splitter.pl --bcfile FILE --prefix PREFIX [--suffix SUFFIX] [--bol|--eol]
[--mismatches N] [--exact] [--partial N] [--help] [--quiet] [--debug]
Arguments:
--bcfile FILE - Barcodes file name. (see explanation below.)
--prefix PREFIX - File prefix. will be added to the output files. Can be used
to specify output directories.
--suffix SUFFIX - File suffix (optional). Can be used to specify file
extensions.
--bol - Try to match barcodes at the BEGINNING of sequences.
(What biologists would call the 5' end, and programmers
would call index 0.)
--eol - Try to match barcodes at the END of sequences.
(What biologists would call the 3' end, and programmers
would call the end of the string.)
NOTE: one of --bol, --eol must be specified, but not both.
--mismatches N - Max. number of mismatches allowed. default is 1.
--exact - Same as '--mismatches 0'. If both --exact and --mismatches
are specified, '--exact' takes precedence.
--partial N - Allow partial overlap of barcodes. (see explanation below.)
(Default is not partial matching)
--quiet - Don't print counts and summary at the end of the run.
(Default is to print.)
--debug - Print lots of useless debug information to STDERR.
--help - This helpful help screen.
Example (Assuming 's_2_100.txt' is a FASTQ file, 'mybarcodes.txt' is
the barcodes file):
$ cat s_2_100.txt | /usr/local/bin/fastx_barcode_splitter.pl --bcfile mybarcodes.txt --bol --mismatches 2 \
--prefix /tmp/bla_ --suffix ".txt"
Barcode file format
-------------------
Barcode files are simple text files. Each line should contain an identifier
(descriptive name for the barcode), and the barcode itself (A/C/G/T),
separated by a TAB character. Example:
#This line is a comment (starts with a 'number' sign)
BC1 GATCT
BC2 ATCGT
BC3 GTGAT
BC4 TGTCT
For each barcode, a new FASTQ file will be created (with the barcode's
identifier as part of the file name). Sequences matching the barcode
will be stored in the appropriate file.
Running the above example (assuming "mybarcodes.txt" contains the above
barcodes), will create the following files:
/tmp/bla_BC1.txt
/tmp/bla_BC2.txt
/tmp/bla_BC3.txt
/tmp/bla_BC4.txt
/tmp/bla_unmatched.txt
The 'unmatched' file will contain all sequences that didn't match any barcode.
Barcode matching
----------------
** Without partial matching:
Count mismatches between the FASTA/Q sequences and the barcodes.
The barcode which matched with the lowest mismatches count (providing the
count is small or equal to '--mismatches N') 'gets' the sequences.
Example (using the above barcodes):
Input Sequence:
GATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
Matching with '--bol --mismatches 1':
GATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (1 mismatch, BC1)
ATCGT (4 mismatches, BC2)
GTGAT (3 mismatches, BC3)
TGTCT (3 mismatches, BC4)
This sequence will be classified as 'BC1' (it has the lowest mismatch count).
If '--exact' or '--mismatches 0' were specified, this sequence would be
classified as 'unmatched' (because, although BC1 had the lowest mismatch count,
it is above the maximum allowed mismatches).
Matching with '--eol' (end of line) does the same, but from the other side
of the sequence.
** With partial matching (very similar to indels):
Same as above, with the following addition: barcodes are also checked for
partial overlap (number of allowed non-overlapping bases is '--partial N').
Example:
Input sequence is ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
(Same as above, but note the missing 'G' at the beginning.)
Matching (without partial overlapping) against BC1 yields 4 mismatches:
ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (4 mismatches)
Partial overlapping would also try the following match:
-ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (1 mismatch)
Note: scoring counts a missing base as a mismatch, so the final
mismatch count is 2 (1 'real' mismatch, 1 'missing base' mismatch).
If running with '--mismatches 2' (meaning allowing upto 2 mismatches) - this
seqeunce will be classified as BC1.
Example: FASTQ Information
Genrating Quality Information on BC54.fq:
$ fastx_quality_stats -i BC54.fq -o bc54_stats.txt
$ fastq_quality_boxplot_graph.sh -i bc54_stats.txt -o bc54_quality.png -t “My Library”
$ fastx_nucleotide_distribution_graph.sh -i bc54_stats.txt -o bc54_nuc.png -t “My Library”
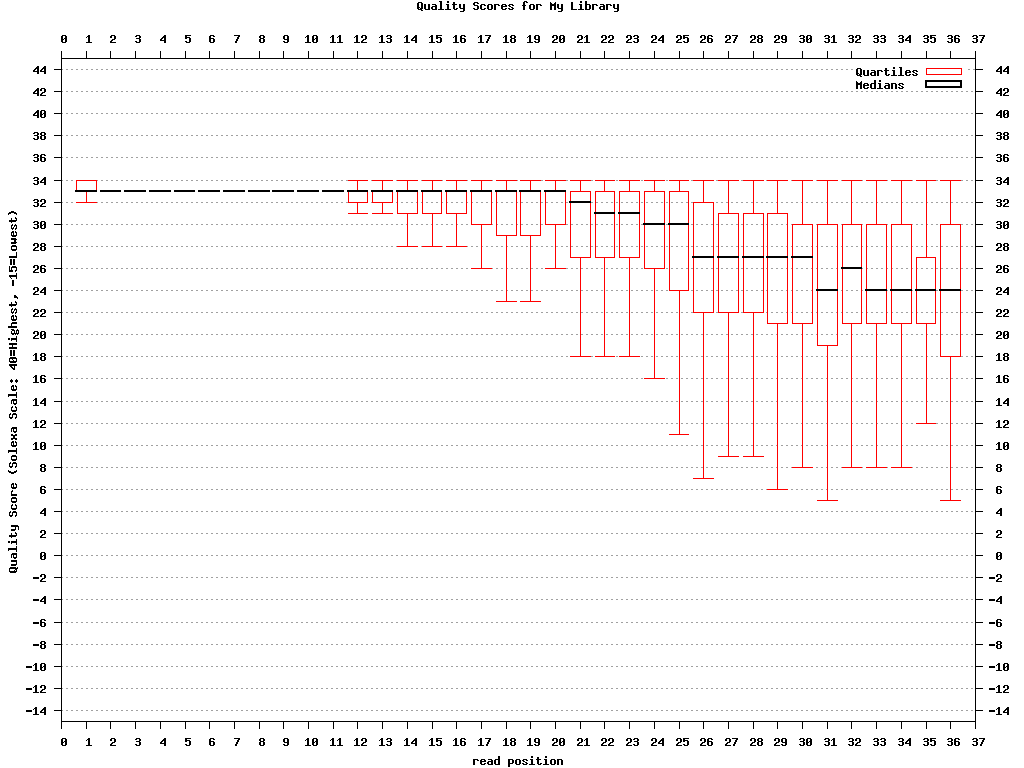
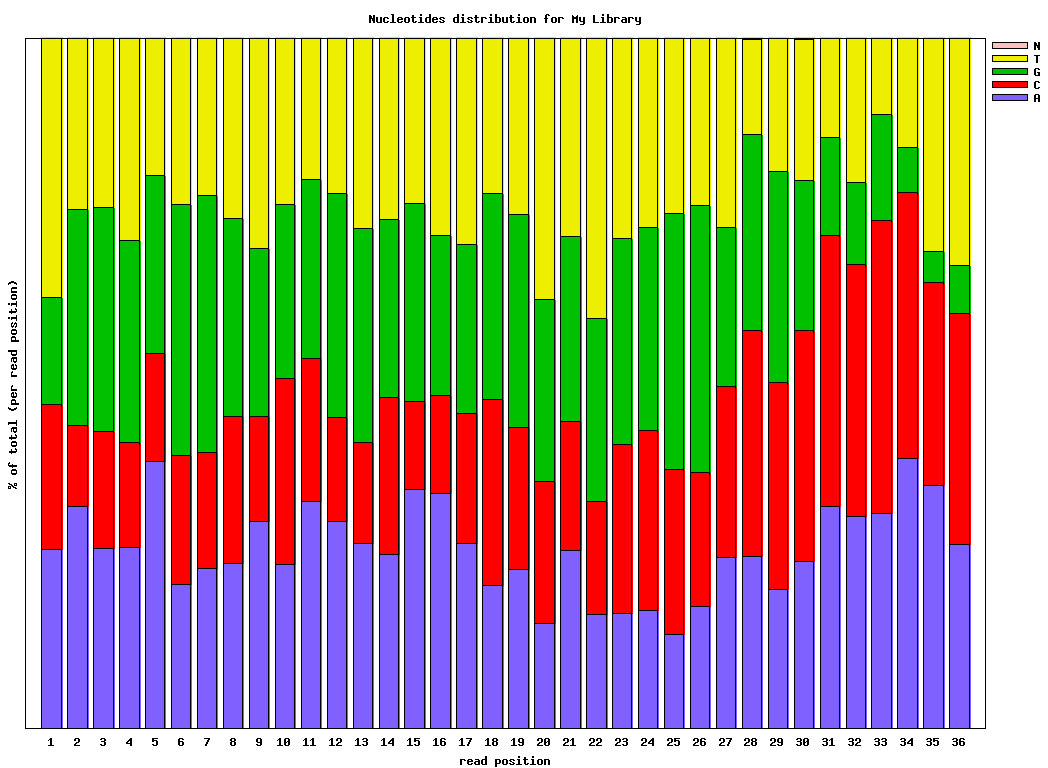
Example: FASTQ/A Manipulation
Common pre-processing work-flow:
Covnerting FASTQ to FASTA
Clipping the Adapter/Linker
Trimming to 27nt (if you’re analyzing miRNAs, for example)
Collapsing the sequences
Plotting the clipping results
Using the FASTX-toolkit from the command line:
1 | $ fastq_to_fasta -v -n -i BC54.fq -o BC54.fa |
Mapping (or any other kind of analysis) of the Clipped + Collapsed FASTA file will be:
quicker - each unique sequence appears only once in the FASTA file.
more accurate - the Adapter/Linker sequence was removed from the 3’ end, and will affect the mapping results.
