概念
宏基因组( Metagenome)(也称微生物环境基因组 Microbial Environmental Genome, 或元基因组) 。定义为”the genomes of the total microbiota found in nature” , 即生境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因, 目前主要指环境样品中的细菌和真菌的基因组总和。
宏基因组学(或元基因组学, metagenomics)就是一种以环境样品中的微生物群体基因组为研究对象, 以功能基因筛选和/或测序分析为研究手段, 以微生物多样性、 种群结构、 进化关系、 功能活性、 相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。
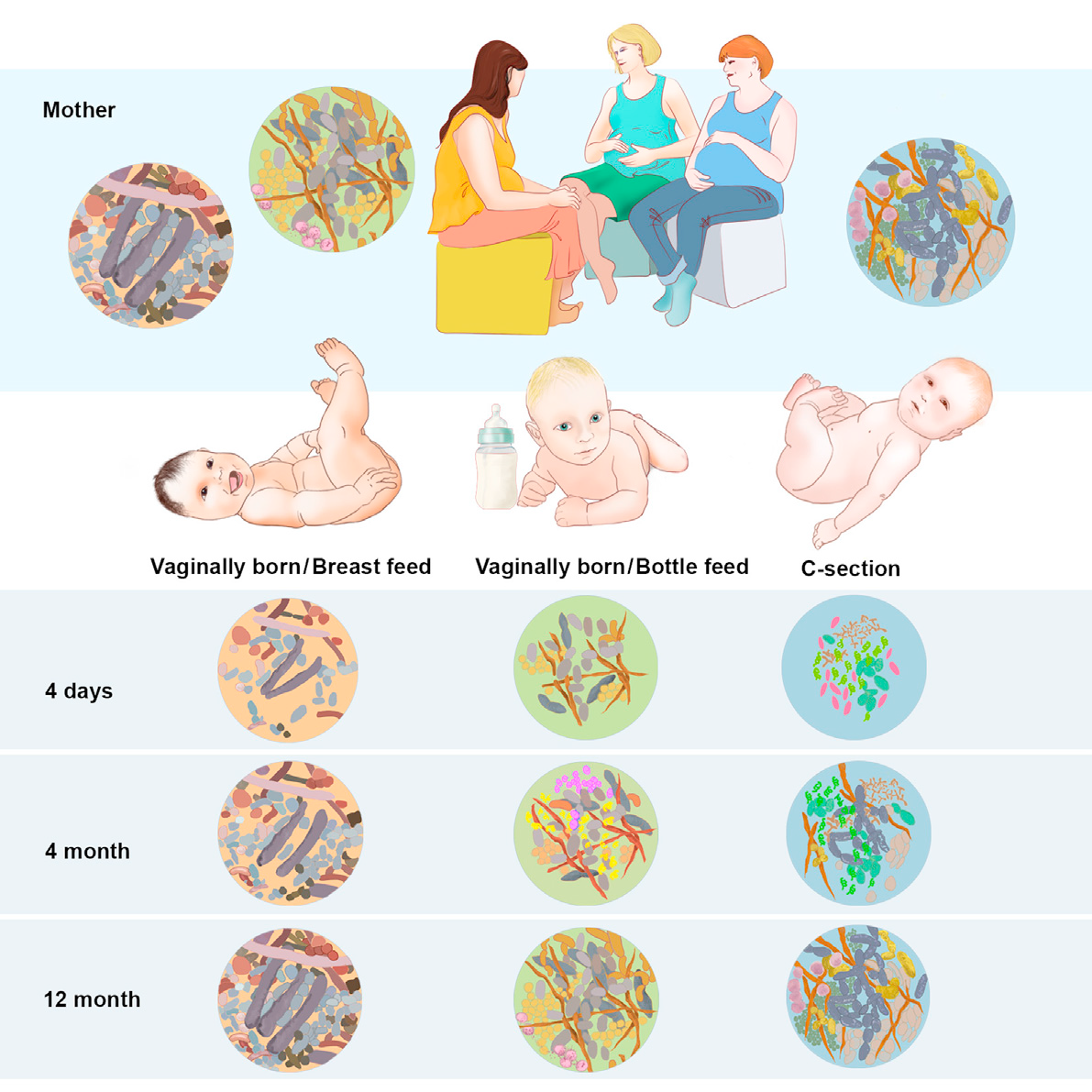
物种丰富度(species richness):用来描述和量化微生物群落,反映特定区域物种的数量。
均匀度(evenness):用来量化一个群体中(少数的优势物种和绝大多数的稀有物种)的不同代表物种,反映各物种个体数目分配的均匀程度。假设存在另外一个群体其含由同之前群体所含物种总数相同的物种,唯一不同的是这些物种均较为常见,即这两个群体拥有相同的物种丰富度,但其某一物种的丰度不同,如何评价这两个群体的多样性?
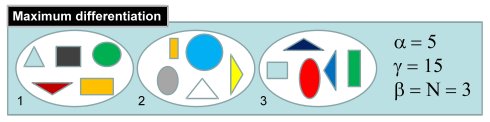
为更好的描述和比较不同群体的多样性,提出适应宏基因组的新度量,α多样性,β多样性和γ多样性,其相互关系为β = γ/α。
宏基因组发展历程
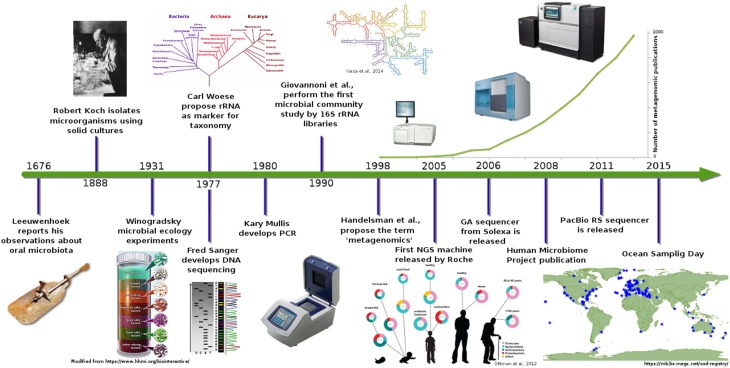
微生物群落研究始于1676年Leeuwenhoek发现第一个微生物,在70年代末 Carl Woese提出16S rRNA基因可用于物种分类,随后几十年分子技术如PCR、FISH、DGGE等的发展促使微生态学研究进入”new uncultured world”,近十年来随着下一代测序技术(NGS)的出现微生物群落研究已经从简单的物种发现发展为宏基因组学研究,即基于NGS技术研究环境样品所包含的全部微生物的遗传组成及其群落功能。
目前,根据测序数据类型的不同,宏基因组测序被分为两类:全基因组测序(full shutgun metagenomics)和扩增子测序(marker gene amplification metagenomics)。
这种策略可以回答以下问题:
1)环境中包含哪些微生物?
2)微生物群落具有哪些功能?
3)微生物间如何相互作用以维持生态平衡?
宏基因组分析流程
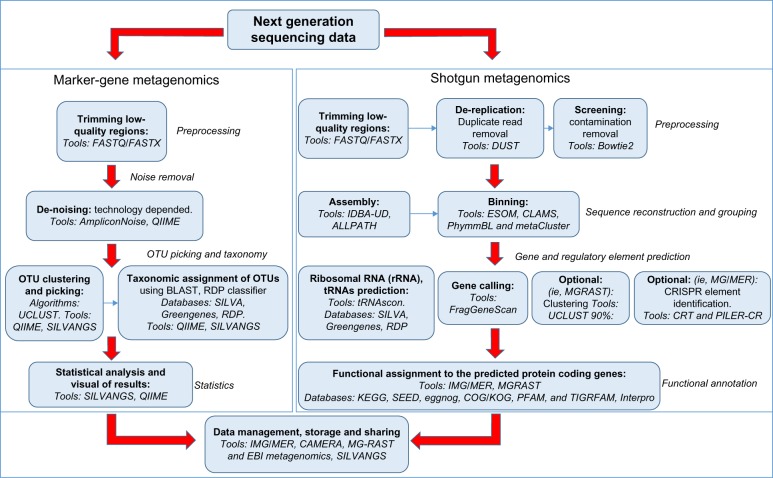
估计宏基因组样本中的物种组成及丰度
宏基因组中的物种分类,一般用OTU (operational taxonomic unit), 即可操作物种单元来表示。在典型情况下,原核生物的OUT使用16S rDNA来衡量,真核生物的OUT使用18s rDNA来衡量。
但选择16S/18S rDNA鉴定物种,存在以下几个问题:1)rDNA之间的平行转移来干扰rDNA鉴定的可靠性。2)在单个细菌中,16r DNA可能存在序列不同的几个拷贝,干扰估计OTU数目的准确性。所以,其他备选的标记基因,比如单拷贝的看家基因被推荐用来作为菌种鉴定的标记。
研究成果
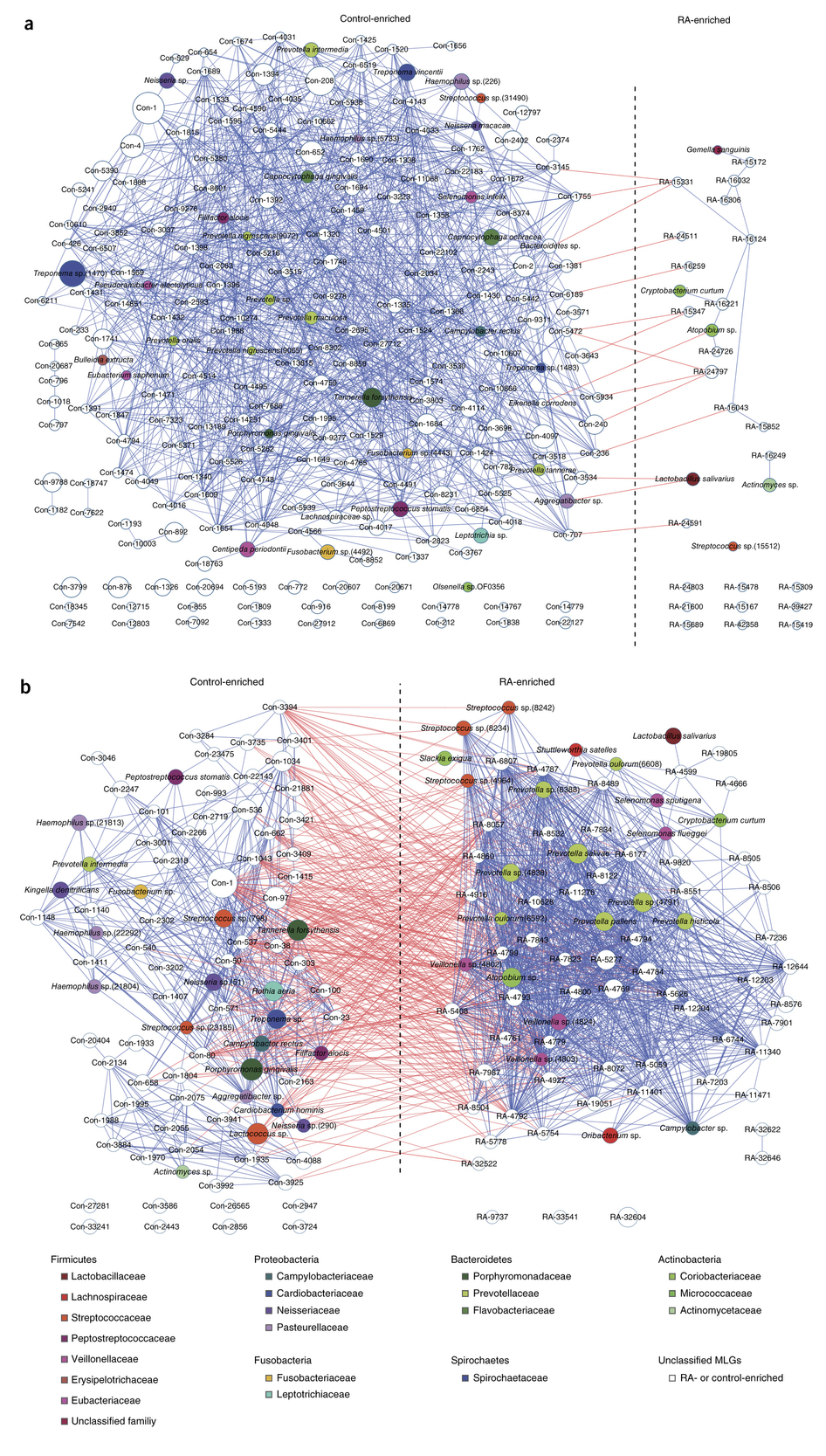
通过宏基因组shotgun测序和宏基因组关联分析来自类风湿性关节炎(RA)患者和健康人体的fecal, dental and salivary样本,观察到中肠和口腔中微生物组的一致性,同时观察到RA患者中肠和口腔中的微生物组失调,经过RA治疗后部分恢复正常;与健康人群相比,RA患者中gut, dental or saliva微生物组显著变化的个体与临床诊断结果密切相关;尤其是嗜血杆菌数量在RA患者的gut, dental and saliva中均下降,与血清自身抗体水平呈负相关;相反唾液乳杆菌在RA患者的gut, dental and saliva中超量存在,并随RA程度增加;功能上,RA患者个体微生物群落在氧化还原环境、铁硫锌离子和精氨酸的转运和代谢上发生变化。
参考资料
The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics
Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies
