Genome_Scripts
A repository for scripts used in genome project ➡ https://tiramisutes.github.io/Genome_Scripts/
Welcome to Genome Scripts
A repository for scripts used in genome project.
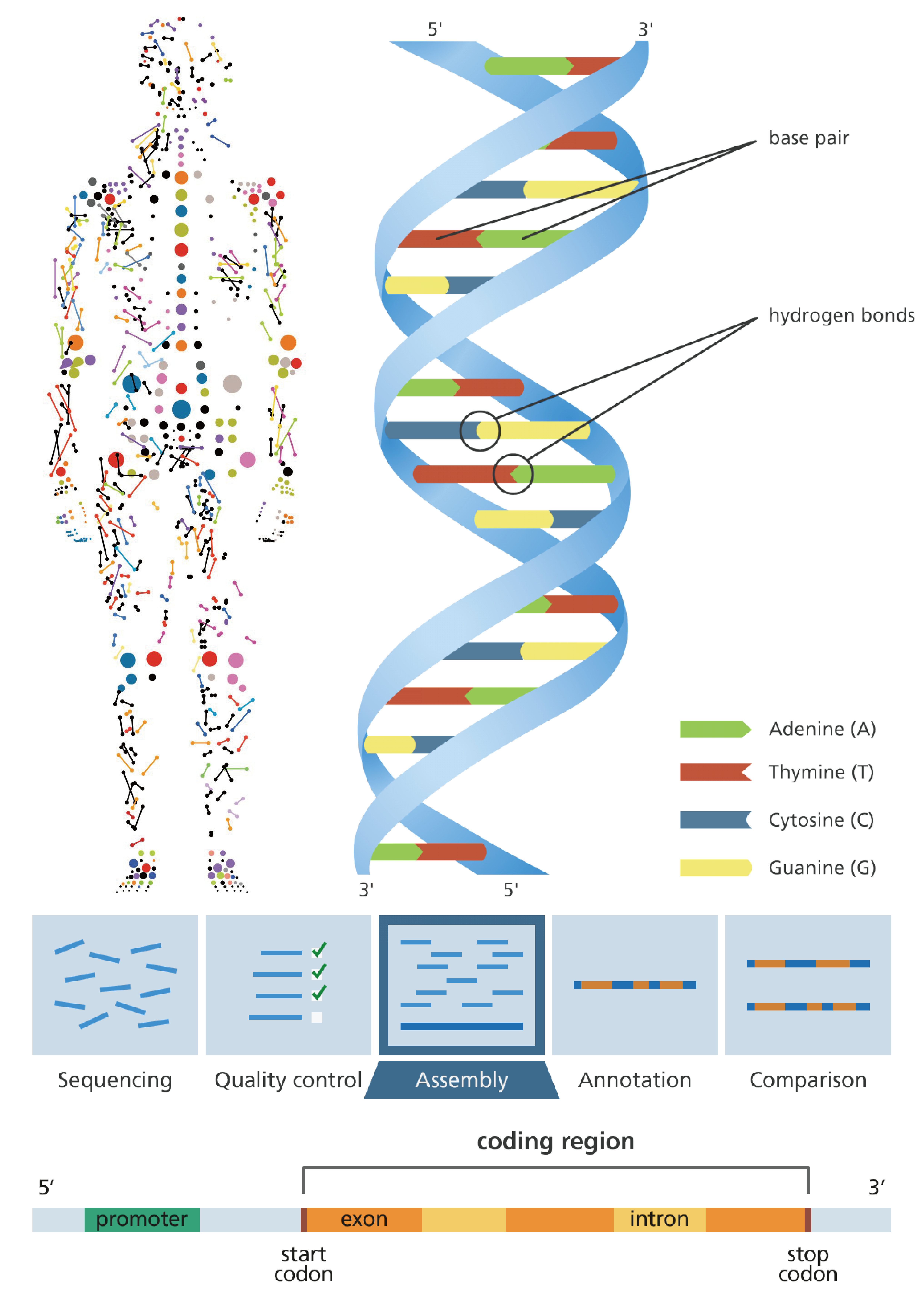
What We can do?
1. Change the genome fa and gff3 format file to Genbank [gff]
python scripts/gff_convert.py -f genbank -s -o /home/zpxu /home/zpxu/genome/annotation.gff3 /home/zpxu/genome/genome.fa
2. Preparing genomes for submission to NCBI [WGS2NCBI]
cd /home/zpxu/software/wgs2ncbi
./script/wgs2ncbi prepare -conf ./share/wgs2ncbi.ini
./script/wgs2ncbi process -conf ./share/wgs2ncbi.ini
./script/wgs2ncbi convert -conf ./share/wgs2ncbi.ini
./script/wgs2ncbi compress -conf ./share/wgs2ncbi.ini
3. Remove duplicate sequence from fasta format files (different IDs but the same sequence) [FASTA]
fasta_unique.pl input.fa >unique.fa 2>unique.tab
removerep.pl input.fa output.fa
4. Some gadget used to process the txt/csv format files [TxtTools]
Combine two files
python combine_files.py -f1 csv -f2 table -L gene -R GeneID -w right -o out.csv file1 file2
5. average read length [FASTQ]
./fastq_stat.sh AS285A_R1.clean.fastq AS285A_R2.clean.fastq