Chrome(Download)是Google公司开发的一个现代化的网页浏览器,作为三大浏览器之一 它搭载了被称为V8的高效率Javascript引擎。
由于简洁的界面风格 和便捷易用的特点 Chrome的市场份额已经升至45% ,一举超越了Internet Explorer和Mozilla Firefox成为全球第一大浏览器。
Chrome(Download)是Google公司开发的一个现代化的网页浏览器,作为三大浏览器之一 它搭载了被称为V8的高效率Javascript引擎。
由于简洁的界面风格 和便捷易用的特点 Chrome的市场份额已经升至45% ,一举超越了Internet Explorer和Mozilla Firefox成为全球第一大浏览器。
CummeRbund was designed to help simplify the analysis and exploration portion of RNA-Seq data derrived from the output of a differential expression analysis using cuffdiff with the goal of providing fast and intuitive access to your results.
$ cd cuffdiff
$ R
> library(cummeRbund)
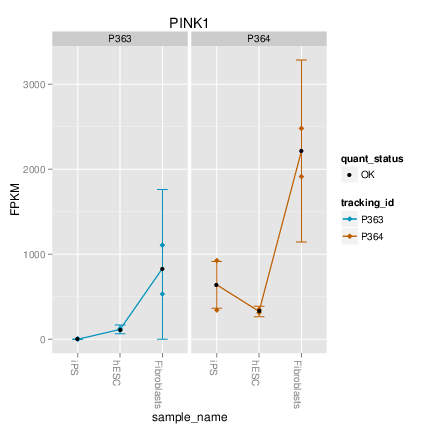
> cuff<-readCufflinks()
> cuff
Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA-Seq samples. It accepts aligned RNA-Seq reads and assembles the alignments into a parsimonious set of transcripts. Cufflinks then estimates the relative abundances of these transcripts based on how many reads support each one, taking into account biases in library preparation protocols.
cufflinks [options] <aligned_reads.(sam/bam)>
for example:
$ cufflinks -p 8 -G transcript.gtf --library-type fr-unstranded -o cufflinks_output tophat_out/accepted_hits.bam
-o/--output-dir write all output files to this directory [ default: ./ ]
-p/--num-threads number of threads used during analysis [ default: 1 ]
--seed value of random number generator seed [ default: 0 ]
-G/--GTF quantitate against reference transcript annotations
-g/--GTF-guide use reference transcript annotation to guide assembly
Use to merge together several Cufflinks assemblies.
cuffmerge [options]* <assembly_GTF_list.txt>
cuffmerge takes several assembly GTF files from Cufflinks’ as input. Input GTF files must be specified in a “manifest” file listing full paths to the files.
<assembly_GTF_list.txt>
Text file “manifest” with a list (one per line) of GTF files that you’d like to merge together into a single GTF file.
Use to find significant changes in transcript expression, splicing, and promoter use.
cuffdiff [options]* <transcripts.gtf> \
<sample1_replicate1.sam[,…,sample1_replicateM.sam]> \
<sample2_replicate1.sam[,…,sample2_replicateM.sam]> … \
[sampleN.sam_replicate1.sam[,…,sample2_replicateM.sam]]
gene_exp.diff Gene-level differential expression. Tests differences in the summed FPKM of transcripts sharing each gene_id
cuffdiff过程中同一处理的多个样本间用逗号分隔,不同处理间空格分隔.
TopHat is a fast splice junction mapper for RNA-Seq reads. It aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify splice junctions between exons.
tophat [options]* [reads1_2,...readsN_2]
for example:
tophat -p 8 -G genes.gtf -o C1_R1_thout genome C1_R1_1.fq C1_R1_2.fq
-p 代表线程
-G 代表转录本注释信息
-o 输出文件夹
--segment-length 25 (将redas分成的最小比对片段)
--segment-mismatches 1 (片段比对错配碱基数)
--library-type (是否链特异性)fr-unstranded
—transcriptome-index (转录本的bowtie-index文件)
比对输出文件:
accepted_hits.bam(比对输出)
junctions.bed
insertions.bed and deletions.bed
Sort is a Linux program used for printing lines of input text files and concatenation of all files in sorted order. Sort command takes blank space as field separator and entire Input file as sort key. It is important to notice that sort command don’t actually sort the files but only print the sorted output, until your redirect the output.
This article aims at deep insight of Linux ‘sort’ command with 14 useful practical examples that will show you how to use sort command in Linux.
The option ‘-e’ in the below command enables interpretion of backslash and /n tells echo to write each string to a new line.
1 | $ echo -e "computer\nmouse\nLAPTOP\ndata\nRedHat\nlaptop\ndebian\nlaptop" > tecmint.txt |
1 | $ cat tecmint.txt |
1 | $ sort tecmint.txt |
Note: The above command don’t actually sort the contents of text file but only show the sorted output on terminal.
1 | $ sort tecmint.txt > sorted.txt |
1 | $ sort -r tecmint.txt > reversesorted.txt |
1 | $ ls -l /home/$USER > /home/$USER/Desktop/tecmint/lsl.txt |
Now will see examples to sort the contents on the basis of other field and not the default initial characters.
1 | $ sort -nk2 lsl.txt |
Note: The ‘-n’ option in the above example sort the contents numerically. Option ‘-n’ must be used when we wanted to sort a file on the basis of a column which contains numerical values.
1 | $ sort -k9 lsl.txt |
1 | $ ls -l /home/$USER | sort -nk5 |
1 | $ cat tecmint.txt |
Rules so far (what we have observed):1
2
3
4Lines starting with numbers are preferred in the list and lies at the top until otherwise specified (-r).
Lines starting with lowercase letters are preferred in the list and lies at the top until otherwise specified (-r).
Contents are listed on the basis of occurrence of alphabets in dictionary until otherwise specified (-r).
Sort command by default treat each line as string and then sort it depending upon dictionary occurrence of alphabets (Numeric preferred; see rule – 1) until otherwise specified.
1 | $ ls -lA /home/$USER > /home/$USER/Desktop/tecmint/lsla.txt |
Those having understanding of ‘ls’ command knows that ‘ls -lA’=’ls -l’ + Hidden files. So most of the contents on these two files would be same.
1 | $ sort lsl.txt lsla.txt |
Notice the repetition of files and folders.
1 | $ sort -u lsl.txt lsla.txt |
Notice that duplicates has been omitted from the output. Also, you can write the output to a new file by redirecting the output to a file.
1 | $ ls -l /home/$USER | sort -t "," -nk2,5 -k9 |
That’s all for now. In the next article we will cover a few more examples of ‘sort’ command in detail for you. Till then stay tuned and connected to Tecmint. Keep sharing. Keep commenting. Like and share us and help us get spread.
More info: sort
Here you’ll find a short description and examples of how to use the FASTX-toolkit from the command line.
Most tools show usage information with -h.
Tools can read from STDIN and write to STDOUT, or
from a specific input file (-i) and specific output file (-o).
Tools can operate silently (producing no output if everything was OK), or
print a short summary (-v).
If output goes to STDOUT, the summary will be printed to STDERR.
If output goes to a file, the summary will be printed to STDOUT.
Some tools can compress the output with GZIP (-z).
1 | $ fastq_to_fasta -h |
$ fastx_quality_stats -h
usage: fastx_quality_stats [-h] [-i INFILE] [-o OUTFILE]
version 0.0.6 (C) 2008 by Assaf Gordon (gordon@cshl.edu)
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
If FASTA file is given, only nucleotides
distribution is calculated (there's no quality info).
[-o OUTFILE] = TEXT output file. default is STDOUT.
The output TEXT file will have the following fields (one row per column):
column = column number (1 to 36 for a 36-cycles read solexa file)
count = number of bases found in this column.
min = Lowest quality score value found in this column.
max = Highest quality score value found in this column.
sum = Sum of quality score values for this column.
mean = Mean quality score value for this column.
Q1 = 1st quartile quality score.
med = Median quality score.
Q3 = 3rd quartile quality score.
IQR = Inter-Quartile range (Q3-Q1).
lW = 'Left-Whisker' value (for boxplotting).
rW = 'Right-Whisker' value (for boxplotting).
A_Count = Count of 'A' nucleotides found in this column.
C_Count = Count of 'C' nucleotides found in this column.
G_Count = Count of 'G' nucleotides found in this column.
T_Count = Count of 'T' nucleotides found in this column.
N_Count = Count of 'N' nucleotides found in this column.
max-count = max. number of bases (in all cycles)
$ fastq_quality_boxplot_graph.sh -h
Solexa-Quality BoxPlot plotter
Generates a solexa quality score box-plot graph
Usage: /usr/local/bin/fastq_quality_boxplot_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "solexa_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title (usually the solexa file name) - will be plotted on the graph.
$ fastx_nucleotide_distribution_graph.sh -h
FASTA/Q Nucleotide Distribution Plotter
Usage: /usr/local/bin/fastx_nucleotide_distribution_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "fastx_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title - will be plotted on the graph.
$ fastx_clipper -h
usage: fastx_clipper [-h] [-a ADAPTER] [-D] [-l N] [-n] [-d N] [-c] [-C] [-o] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-a ADAPTER] = ADAPTER string. default is CCTTAAGG (dummy adapter).
[-l N] = discard sequences shorter than N nucleotides. default is 5.
[-d N] = Keep the adapter and N bases after it.
(using '-d 0' is the same as not using '-d' at all. which is the default).
[-c] = Discard non-clipped sequences (i.e. - keep only sequences which contained the adapter).
[-C] = Discard clipped sequences (i.e. - keep only sequences which did not contained the adapter).
[-k] = Report Adapter-Only sequences.
[-n] = keep sequences with unknown (N) nucleotides. default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-D] = DEBUG output.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
$ fastx_renamer -h
usage: fastx_renamer [-n TYPE] [-h] [-z] [-v] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.10 by A. Gordon (gordon@cshl.edu)
[-n TYPE] = rename type:
SEQ - use the nucleotides sequence as the name.
COUNT - use simply counter as the name.
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
$ fastx_trimmer -h
usage: fastx_trimmer [-h] [-f N] [-l N] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-f N] = First base to keep. Default is 1 (=first base).
[-l N] = Last base to keep. Default is entire read.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
$ fastx_collapser -h
usage: fastx_collapser [-h] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-v] = verbose: print short summary of input/output counts
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
$ fastx_artifacts_filter -h
usage: fastq_artifacts_filter [-h] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-z] = Compress output with GZIP.
[-v] = Verbose - report number of processed reads.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
$ fastq_quality_filter -h
usage: fastq_quality_filter [-h] [-v] [-q N] [-p N] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-q N] = Minimum quality score to keep.
[-p N] = Minimum percent of bases that must have [-q] quality.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
$ fastx_reverse_complement -h
usage: fastx_reverse_complement [-h] [-r] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
$ fasta_formatter -h
usage: fasta_formatter [-h] [-i INFILE] [-o OUTFILE] [-w N] [-t] [-e]
Part of FASTX Toolkit 0.0.7 by gordon@cshl.edu
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-w N] = max. sequence line width for output FASTA file.
When ZERO (the default), sequence lines will NOT be wrapped -
all nucleotides of each sequences will appear on a single
line (good for scripting).
[-t] = Output tabulated format (instead of FASTA format).
Sequence-Identifiers will be on first column,
Nucleotides will appear on second column (as single line).
[-e] = Output empty sequences (default is to discard them).
Empty sequences are ones who have only a sequence identifier,
but not actual nucleotides.
Input Example:
>MY-ID
AAAAAGGGGG
CCCCCTTTTT
AGCTN
Output example with unlimited line width [-w 0]:
>MY-ID
AAAAAGGGGGCCCCCTTTTTAGCTN
Output example with max. line width=7 [-w 7]:
>MY-ID
AAAAAGG
GGGTTTT
TCCCCCA
GCTN
Output example with tabular output [-t]:
MY-ID AAAAAGGGGGCCCCCTTTTAGCTN
example of empty sequence:
(will be discarded unless [-e] is used)
>REGULAR-SEQUENCE-1
AAAGGGTTTCCC
>EMPTY-SEQUENCE
>REGULAR-SEQUENCE-2
AAGTAGTAGTAGTAGT
GTATTTTATAT
$ fasta_nucleotide_changer -h
usage: fasta_nucleotide_changer [-h] [-z] [-v] [-i INFILE] [-o OUTFILE] [-r] [-d]
version 0.0.7
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-v] = Verbose mode. Prints a short summary.
with [-o], summary is printed to STDOUT.
Otherwise, summary is printed to STDERR.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-r] = DNA-to-RNA mode - change T's into U's.
[-d] = RNA-to-DNA mode - change U's into T's.
$ fasta_clipping_histogram.pl
Create a Linker Clipping Information Histogram
usage: fasta_clipping_histogram.pl INPUT_FILE.FA OUTPUT_FILE.PNG
INPUT_FILE.FA = input file (in FASTA format, can be GZIPped)
OUTPUT_FILE.PNG = histogram image
$ fastx_barcode_splitter.pl
Barcode Splitter, by Assaf Gordon (gordon@cshl.edu), 11sep2008
This program reads FASTA/FASTQ file and splits it into several smaller files,
Based on barcode matching.
FASTA/FASTQ data is read from STDIN (format is auto-detected.)
Output files will be writen to disk.
Summary will be printed to STDOUT.
usage: /usr/local/bin/fastx_barcode_splitter.pl --bcfile FILE --prefix PREFIX [--suffix SUFFIX] [--bol|--eol]
[--mismatches N] [--exact] [--partial N] [--help] [--quiet] [--debug]
Arguments:
--bcfile FILE - Barcodes file name. (see explanation below.)
--prefix PREFIX - File prefix. will be added to the output files. Can be used
to specify output directories.
--suffix SUFFIX - File suffix (optional). Can be used to specify file
extensions.
--bol - Try to match barcodes at the BEGINNING of sequences.
(What biologists would call the 5' end, and programmers
would call index 0.)
--eol - Try to match barcodes at the END of sequences.
(What biologists would call the 3' end, and programmers
would call the end of the string.)
NOTE: one of --bol, --eol must be specified, but not both.
--mismatches N - Max. number of mismatches allowed. default is 1.
--exact - Same as '--mismatches 0'. If both --exact and --mismatches
are specified, '--exact' takes precedence.
--partial N - Allow partial overlap of barcodes. (see explanation below.)
(Default is not partial matching)
--quiet - Don't print counts and summary at the end of the run.
(Default is to print.)
--debug - Print lots of useless debug information to STDERR.
--help - This helpful help screen.
Example (Assuming 's_2_100.txt' is a FASTQ file, 'mybarcodes.txt' is
the barcodes file):
$ cat s_2_100.txt | /usr/local/bin/fastx_barcode_splitter.pl --bcfile mybarcodes.txt --bol --mismatches 2 \
--prefix /tmp/bla_ --suffix ".txt"
Barcode file format
-------------------
Barcode files are simple text files. Each line should contain an identifier
(descriptive name for the barcode), and the barcode itself (A/C/G/T),
separated by a TAB character. Example:
#This line is a comment (starts with a 'number' sign)
BC1 GATCT
BC2 ATCGT
BC3 GTGAT
BC4 TGTCT
For each barcode, a new FASTQ file will be created (with the barcode's
identifier as part of the file name). Sequences matching the barcode
will be stored in the appropriate file.
Running the above example (assuming "mybarcodes.txt" contains the above
barcodes), will create the following files:
/tmp/bla_BC1.txt
/tmp/bla_BC2.txt
/tmp/bla_BC3.txt
/tmp/bla_BC4.txt
/tmp/bla_unmatched.txt
The 'unmatched' file will contain all sequences that didn't match any barcode.
Barcode matching
----------------
** Without partial matching:
Count mismatches between the FASTA/Q sequences and the barcodes.
The barcode which matched with the lowest mismatches count (providing the
count is small or equal to '--mismatches N') 'gets' the sequences.
Example (using the above barcodes):
Input Sequence:
GATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
Matching with '--bol --mismatches 1':
GATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (1 mismatch, BC1)
ATCGT (4 mismatches, BC2)
GTGAT (3 mismatches, BC3)
TGTCT (3 mismatches, BC4)
This sequence will be classified as 'BC1' (it has the lowest mismatch count).
If '--exact' or '--mismatches 0' were specified, this sequence would be
classified as 'unmatched' (because, although BC1 had the lowest mismatch count,
it is above the maximum allowed mismatches).
Matching with '--eol' (end of line) does the same, but from the other side
of the sequence.
** With partial matching (very similar to indels):
Same as above, with the following addition: barcodes are also checked for
partial overlap (number of allowed non-overlapping bases is '--partial N').
Example:
Input sequence is ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
(Same as above, but note the missing 'G' at the beginning.)
Matching (without partial overlapping) against BC1 yields 4 mismatches:
ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (4 mismatches)
Partial overlapping would also try the following match:
-ATTTACTATGTAAAGATAGAAGGAATAAGGTGAAG
GATCT (1 mismatch)
Note: scoring counts a missing base as a mismatch, so the final
mismatch count is 2 (1 'real' mismatch, 1 'missing base' mismatch).
If running with '--mismatches 2' (meaning allowing upto 2 mismatches) - this
seqeunce will be classified as BC1.
$ fastx_quality_stats -i BC54.fq -o bc54_stats.txt
$ fastq_quality_boxplot_graph.sh -i bc54_stats.txt -o bc54_quality.png -t “My Library”
$ fastx_nucleotide_distribution_graph.sh -i bc54_stats.txt -o bc54_nuc.png -t “My Library”
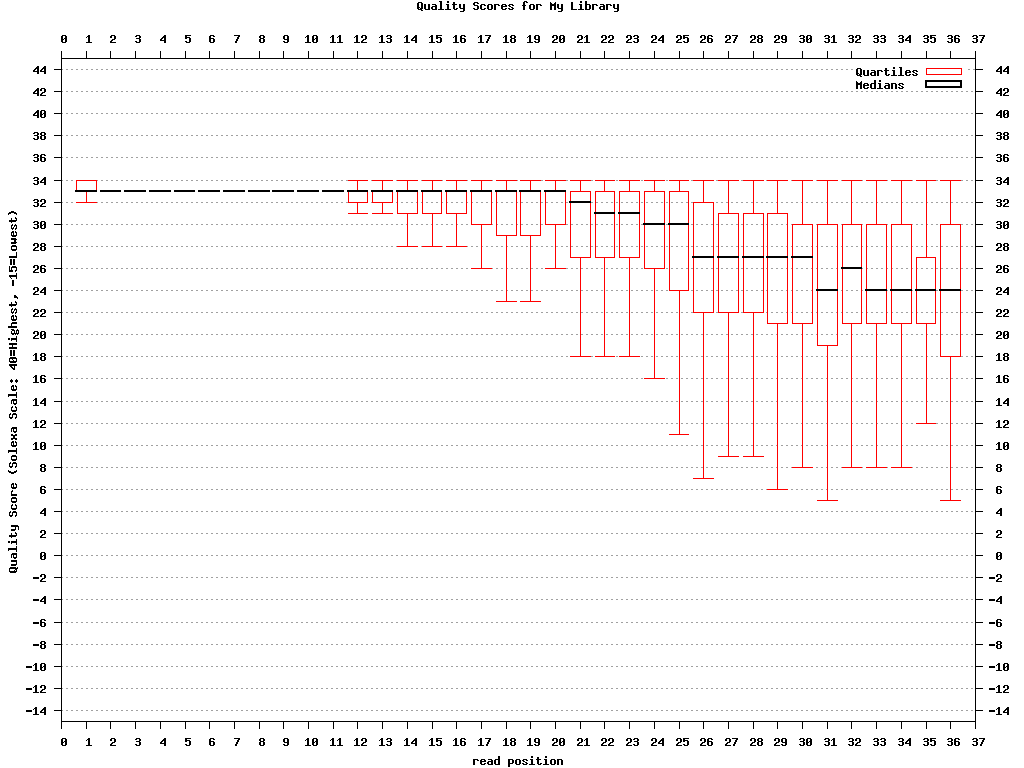
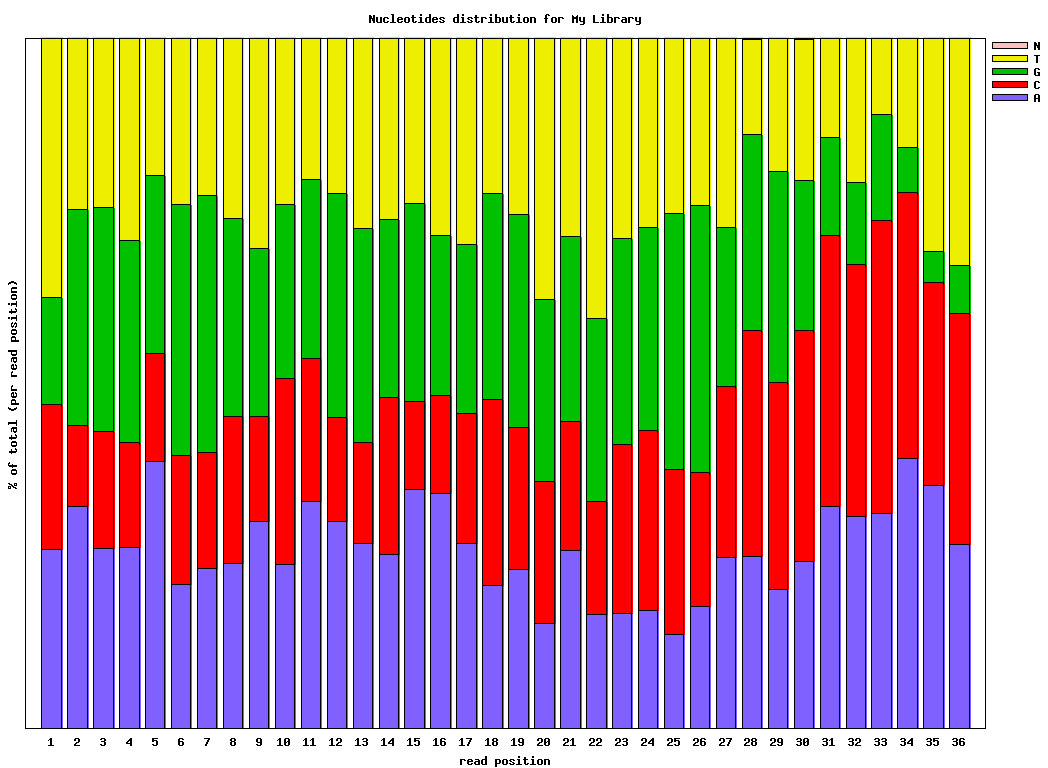
Covnerting FASTQ to FASTA
Clipping the Adapter/Linker
Trimming to 27nt (if you’re analyzing miRNAs, for example)
Collapsing the sequences
Plotting the clipping results
1 | $ fastq_to_fasta -v -n -i BC54.fq -o BC54.fa |
quicker - each unique sequence appears only once in the FASTA file.
more accurate - the Adapter/Linker sequence was removed from the 3’ end, and will affect the mapping results.
Welcome to Hexo! This is your very first post. Check documentation for more info. If you get any problems when using Hexo, you can find the answer in troubleshooting or you can ask me on GitHub.
1 | $ hexo new "My New Post" |
More info: Writing
1 | $ hexo server |
More info: Server
1 | $ hexo generate |
More info: Generating
1 | $ hexo deploy |
More info: Deployment
Let us start new year with these Unix command line tricks to increase productivity at the Terminal. I have found them over the years and I’m now going to share with you.

I had a huge log file 200GB I need to delete on a production web server. My rm and ls command was crashed and I was afraid that the system to a crawl with huge disk I/O load. To remove a HUGE file, enter:
1 | > /path/to/file.log |
Try the script command line utility to create a typescript of everything printed on your terminal.
1 | script my.terminal.session |
Type commands:1
2
3ls
date
sudo service foo stop
To exit (to end script session) type exit or logout or press control-D1
exit
To view type:
1 | more my.terminal.session |
As my journey continues with Linux and Unix shell, I made a few mistakes. I accidentally deleted /tmp folder. To restore it all you have to do is:1
2
3
4mkdir /tmp
chmod 1777 /tmp
chown root:root /tmp
ls -ld /tmp
For privacy of my data I wanted to lock down /downloads on my file server. So I ran:1
chmod 0000 /downloads
The root user can still has access and ls and cd commands will not work. To go back:1
chmod 0755 /downloads
Afraid that root user or someone may snoop into your personal text files? Try password protection to a file in vim, type:1
2
3
4
5
6
7
8
9
10vim +X filename
```
Or, before quitting in vim use :X vim command to encrypt your file and vim will prompt for a password.
### Clear gibberish all over the screen
Just type:
``` bash
reset
Pass the -h or -H (and other options) command line option to GNU or BSD utilities to get output of command commands like ls, df, du, in human-understandable formats:
1 | ls -lh |
Just type:1
2
3
4
5## linux version ##
lslogins
## BSD version ##
logins
Sample outputs:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29UID USER PWD-LOCK PWD-DENY LAST-LOGIN GECOS
0 root 0 0 22:37:59 root
1 bin 0 1 bin
2 daemon 0 1 daemon
3 adm 0 1 adm
4 lp 0 1 lp
5 sync 0 1 sync
6 shutdown 0 1 2014-Dec17 shutdown
7 halt 0 1 halt
8 mail 0 1 mail
10 uucp 0 1 uucp
11 operator 0 1 operator
12 games 0 1 games
13 gopher 0 1 gopher
14 ftp 0 1 FTP User
27 mysql 0 1 MySQL Server
38 ntp 0 1
48 apache 0 1 Apache
68 haldaemon 0 1 HAL daemon
69 vcsa 0 1 virtual console memory owner
72 tcpdump 0 1
74 sshd 0 1 Privilege-separated SSH
81 dbus 0 1 System message bus
89 postfix 0 1
99 nobody 0 1 Nobody
173 abrt 0 1
497 vnstat 0 1 vnStat user
498 nginx 0 1 nginx user
499 saslauth 0 1 "Saslauthd user"
So I accidentally untar a tarball in /var/www/html/ directory instead of /home/projects/www/current. It created mess in /var/www/html/. The easiest way to fix this mess:
1 | cd /var/www/html/ |
Seriously, you need to try out htop instead of top:1
sudo htop
Just type !!. For example:
1 | /myhome/dir/script/name arg1 arg2 |
The !! repeats the most recent command. To run the most recent command beginning with “foo”:1
2
3!foo
# Run the most recent command beginning with "service" as root
sudo !service
The !$ use to run command with the last argument of the most recent command:1
2
3
4
5
6
7
8
9# Edit nginx.conf
sudo vi /etc/nginx/nginx.conf
# Test nginx.conf for errors
/sbin/nginx -t -c /etc/nginx/nginx.conf
# After testing a file with "/sbin/nginx -t -c /etc/nginx/nginx.conf", you
# can edit file again with vi
sudo vi !$
If you need a reminder to leave your terminal, type the following command:
1 | leave +hhmm |
Where,
hhmm - The time of day is in the form hhmm where hh is a time in hours (on a 12 or 24 hour clock), and mm are minutes. All times are converted to a 12 hour clock, and assumed to be in the next 12 hours.
Want to go the directory you were just in? Run:1
cd -
Need to quickly return to your home directory? Enter:1
cd
The variable CDPATH defines the search path for the directory containing directories:1
export CDPATH=/var/www:/nas10
Now, instead of typing cd /var/www/html/ I can simply type the following to cd into /var/www/html path:1
cd html
To edit a file being viewed with less pager, press v. You will have the file for edit under $EDITOR:
1 | less *.c |
Creating a shell function is left as an exercise for the reader
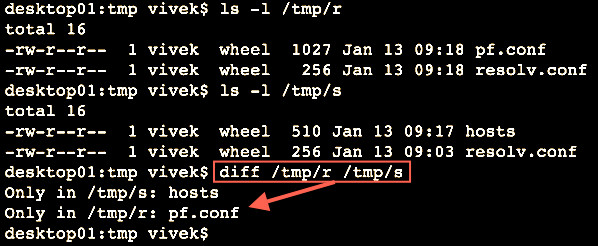
The diff command compare files line by line. It can also compare two directories:1
2
3
4ls -l /tmp/r
ls -l /tmp/s
# Compare two folders using diff ##
diff /tmp/r/ /tmp/s/
You can reformat each paragraph with fmt command. In this example, I’m going to reformat file by wrapping overlong lines and filling short lines:1
fmt file.txt
You can also split long lines, but do not refill i.e. wrap overlong lines, but do not fill short lines:1
fmt -s file.txt
Use the tee command as follows to see the output on screen and also write to a log file named my.log:1
mycoolapp arg1 arg2 input.file | tee my.log
The tee command ensures that you will see mycoolapp output on on the screen and to a file same time.
More info: Unix Command