在转录组数据处理过程中我们经常会用到差异表达分析这一概念,通过比较不同处理或不同组织间基因表达量(FPKM)差异来寻找特异基因,但这前提是你的不同处理或不同组织样本较少,当不同处理或组织有较多样本,如40个,此时的两两比较有780组比较^_^,这根本不是我们想要的结果;
此时就需要WGCNA(weighted gene co-expression network analysis)将复杂的数据进行归纳整理。除了这种最常见的比较差异表达,我们还想知道在不同处理或不同组织间是否有些基因的表达存在内在的联系或相关性?WGCNA同样可以帮助我们预测基因间的相互作用关系。
WGCNA is based on correlation and not differential expression comparisons.
WGCNA术语
权重(weghted)
Module
模块(module):表达模式相似的基因分为一类,这样的一类基因成为模块;
Eigengene
Eigengene(eigen- + gene):基因和样本构成的矩阵,https://en.wiktionary.org/wiki/eigengene;
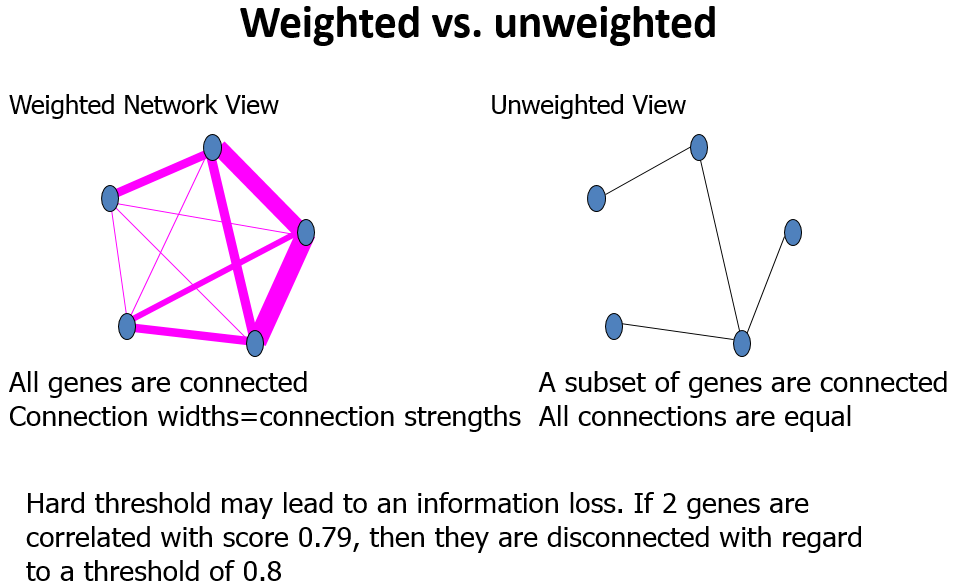
Adjacency Matrix
邻近矩阵:是图的一种存储形式,用一个一维数组存放图中所有顶点数据;用一个二维数组存放顶点间关系(边或弧)的数据,这个二维数组称为邻接矩阵;
Topological Overlap Matrix(TOM)

整体思路
分析构建的网络寻找以下有用信息
R脚本
输入数据为RNA-seq不同处理或组织所有样本的FPKM值组成的矩阵,切记含有 0 的要去掉;1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375setwd("F:/WGCNA")
library(WGCNA)
options(stringsAsFactors = FALSE)
enableWGCNAThreads()
#############################################################################
####################### 一、 数据读入,处理和保存 ##############################
#############################################################################
fpkm<- read.csv("trans_counts.counts.matrix.TMM_normalized.FPKM.nozero.csv")
#~~~~~~~~~~~~~~~~~~
head(fpkm)
GeneID PN_1_TPM PN_1_07_TPM PN_1_08_TPM PN_2_TPM PN_2_09_TPM PN_2_10_TPM
1 MSTRG.1 0.000000 1.456143 1.093308 0.204315 0.000000 0.000000
2 MSTRG.2 1.516181 0.849313 2.010783 1.567867 2.045446 2.246402
3 MSTRG.3 1.305084 1.207246 0.889166 1.470162 0.340003 0.421222
4 MSTRG.4 2.744250 2.791988 2.500786 2.719017 1.954149 2.468110
5 MSTRG.5 1.946825 1.470012 1.263171 0.205806 1.644505 1.638583
6 MSTRG.6 1.325277 0.793530 1.932236 1.210156 1.834274 2.153466
#~~~~~~~~~~~~~~~~~~
dim(fpkm)
names(fpkm)
datExpr0=as.data.frame(t(fpkm[,-c(1)]));
names(datExpr0)=fpkm$trans;
rownames(datExpr0)=names(fpkm)[-c(1)];
#data<-log10(date[,-1]+0.01)
# *************************************************************
# 检测输入基因是否含有缺失值,并处理 ******************************
# *************************************************************
gsg = goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK
#~~~~~~~~~~~~~~~~~~
# 如果上一步返回TRUE则跳过此步,如果返回FALSE则执行如下if语句去掉存在较多缺失值的基因所在行
if (!gsg$allOK)
{
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# Remove the offending genes and samples from the data:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
# 再次检测
dim(datExpr0)
gsg = goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK
#~~~~~~~~~~~~~~~~~~
# ***************************************************************
# 聚类检测输入样本是否含有异常值(obvious outliers),并处理 ********
# ***************************************************************
sampleTree = hclust(dist(datExpr0), method = "average")
#sizeGrWindow(12,9)
par(cex = 0.6)
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
abline(h = 80000, col = "red");
clust = cutreeStatic(sampleTree, cutHeight = 80000, minSize = 10)
table(clust)
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
save(datExpr, file = "AS-green-FPKM-01-dataInput.RData")
#############################################################################
############################ 二、 选择合适的阀值 ##############################
#############################################################################
powers = c(c(1:10), seq(from = 12, to=20, by=2))
# Call the network topology analysis function
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
# Plot the results:
##sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
#############################################################################
############################ 三、 网络构建和可视化 #############################
#############################################################################
#=====================================================================================
#=====================================================================================
# 网络构建有两种方法,One-step和Step-by-step;
# 第一种:一步法进行网络构建
#=====================================================================================
#=====================================================================================
################################################################################
### 3.1 一步法网络构建:One-step network construction and module detection ######
#3.1.1. 网络构建 ###############################################################
###############################################################################
dim(datExpr)
net = blockwiseModules(datExpr, power = 6, maxBlockSize = 6000,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "AS-green-FPKM-TOM",
verbose = 3)
table(net$colors)
###############################################################################
#3.1.2. 绘画结果展示 ###########################################################
###############################################################################
# open a graphics window
#sizeGrWindow(12, 9)
# Convert labels to colors for plotting
mergedColors = labels2colors(net$colors)
# Plot the dendrogram and the module colors underneath
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
###############################################################################
#3.1.3. 结果保存 ###############################################################
###############################################################################
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
table(moduleColors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(MEs, moduleLabels, moduleColors, geneTree,
file = "AS-green-FPKM-02-networkConstruction-auto.RData")
###############################################################################
#3.1.4. 导出网络到 Cytoscape ###################################################
###############################################################################
# Recalculate topological overlap if needed
TOM = TOMsimilarityFromExpr(datExpr, power = 6);
# Read in the annotation file
# annot = read.csv(file = "GeneAnnotation.csv");
# Select modules需要修改,根据table(moduleColors)的结果选择需要导出的模块颜色
modules = c("turquoise", "blue");
# Select module probes选择模块探测
probes = names(datExpr)
inModule = is.finite(match(moduleColors, modules));
modProbes = probes[inModule];
#modGenes = annot$gene_symbol[match(modProbes, annot$substanceBXH)];
# Select the corresponding Topological Overlap
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
# Export the network into edge and node list files Cytoscape can read
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("AS-green-FPKM-One-step-CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
nodeFile = paste("AS-green-FPKM-One-step-CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
#altNodeNames = modGenes,
nodeAttr = moduleColors[inModule]);
#################################################################################################
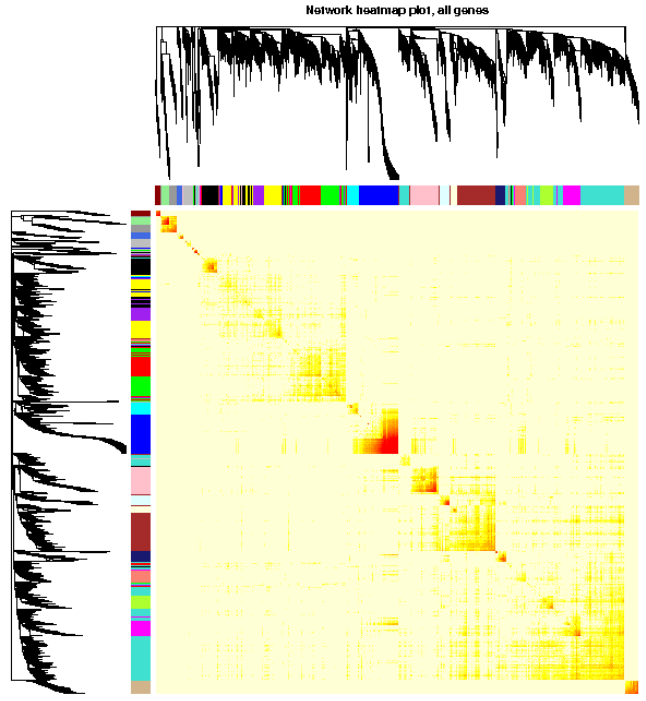
#3.1.5. 分析网络可视化,用heatmap可视化权重网络,heatmap每一行或列对应一个基因,颜色越深表示有较高的邻近
#################################################################################################
options(stringsAsFactors = FALSE);
lnames = load(file = "AS-green-FPKM-01-dataInput.RData");
lnames
lnames = load(file = "AS-green-FPKM-02-networkConstruction-auto.RData");
lnames
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
#====================================
#3.1.5.1. 可视化全部基因网络 ==========
#====================================
# Calculate topological overlap anew: this could be done more efficiently by saving the TOM
# calculated during module detection, but let us do it again here.
dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6);
# Transform dissTOM with a power to make moderately strong connections more visible in the heatmap
plotTOM = dissTOM^7;
# Set diagonal to NA for a nicer plot
diag(plotTOM) = NA;
# Call the plot function
#sizeGrWindow(9,9)
TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")
#====================================
#3.1.5.2. 随便选取1000个基因来可视化 ==
#====================================
nSelect = 1000
# For reproducibility, we set the random seed
set.seed(10);
select = sample(nGenes, size = nSelect);
selectTOM = dissTOM[select, select];
# There's no simple way of restricting a clustering tree to a subset of genes, so we must re-cluster.
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select];
# Open a graphical window
#sizeGrWindow(9,9)
# Taking the dissimilarity to a power, say 10, makes the plot more informative by effectively changing
# the color palette; setting the diagonal to NA also improves the clarity of the plot
plotDiss = selectTOM^7;
diag(plotDiss) = NA;
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes")
#=====================================================================================
#=====================================================================================
# 第二种:一步步的进行网络构建
#=====================================================================================
#=====================================================================================
###############################################################################
### 3.2 Step-by-step network construction and module detection ################
###############################################################################
#2.选择合适的阀值,同上
###############################################################################
#3.2.1. 网络构建 ###############################################################
###############################################################################
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# (1) Co-expression similarity and adjacency ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
softPower = 6;
adjacency = adjacency(datExpr, power = softPower);
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#(2) 邻近矩阵到拓扑矩阵的转换,Turn adjacency into topological overlap ~~~~~
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
TOM = TOMsimilarity(adjacency);
dissTOM = 1-TOM
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# (3) 聚类拓扑矩阵 ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#Call the hierarchical clustering function
geneTree = hclust(as.dist(dissTOM), method = "average");
# Plot the resulting clustering tree (dendrogram)
#sizeGrWindow(12,9)
plot(geneTree, xlab="", sub="", main = "Gene clustering on TOM-based dissimilarity",
labels = FALSE, hang = 0.04);
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#(4) 聚类分支的休整dynamicTreeCut ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# We like large modules, so we set the minimum module size relatively high:
minModuleSize = 30;
# Module identification using dynamic tree cut:
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM,
deepSplit = 2, pamRespectsDendro = FALSE,
minClusterSize = minModuleSize);
table(dynamicMods)
###############################################################################
#3.2.2. 绘画结果展示 ###########################################################
###############################################################################
# Convert numeric lables into colors
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)
# Plot the dendrogram and colors underneath
#sizeGrWindow(8,6)
plotDendroAndColors(geneTree, dynamicColors, "Dynamic Tree Cut",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05,
main = "Gene dendrogram and module colors")
###############################################################################
#3.2.3. 聚类结果相似模块的融合,Merging of modules whose expression profiles are very similar
###############################################################################
#在聚类树中每一leaf是一个短线,代表一个基因,
#不同分之间靠的越近表示有高的共表达基因,将共表达极其相似的modules进行融合
###############################################################################
# Calculate eigengenes
MEList = moduleEigengenes(datExpr, colors = dynamicColors)
MEs = MEList$eigengenes
# Calculate dissimilarity of module eigengenes
MEDiss = 1-cor(MEs);
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average");
# Plot the result
#sizeGrWindow(7, 6)
plot(METree, main = "Clustering of module eigengenes",
xlab = "", sub = "")
#选择有75%相关性的进行融合
MEDissThres = 0.25
# Plot the cut line into the dendrogram
abline(h=MEDissThres, col = "red")
# Call an automatic merging function
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)
# The merged module colors
mergedColors = merge$colors;
# Eigengenes of the new merged modules:
mergedMEs = merge$newMEs;
#绘制融合前(Dynamic Tree Cut)和融合后(Merged dynamic)的聚类图
#sizeGrWindow(12, 9)
#pdf(file = "Plots/geneDendro-3.pdf", wi = 9, he = 6)
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors),
c("Dynamic Tree Cut", "Merged dynamic"),
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
#dev.off()
# 只是绘制融合后聚类图
plotDendroAndColors(geneTree,mergedColors,"Merged dynamic",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
###############################################################################
#3.2.4. 结果保存 ###############################################################
###############################################################################
# Rename to moduleColors
moduleColors = mergedColors
# Construct numerical labels corresponding to the colors
colorOrder = c("grey", standardColors(50));
moduleLabels = match(moduleColors, colorOrder)-1;
MEs = mergedMEs;
# Save module colors and labels for use in subsequent parts
save(MEs, moduleLabels, moduleColors, geneTree, file = "AS-green-FPKM-02-networkConstruction-stepByStep.RData")
###############################################################################
#3.2.5. 导出网络到Cytoscape ####################################################
###############################################################################
# Recalculate topological overlap if needed
TOM = TOMsimilarityFromExpr(datExpr, power = 6);
# Read in the annotation file
# annot = read.csv(file = "GeneAnnotation.csv");
# Select modules需要修改
modules = c("brown", "red");
# Select module probes
probes = names(datExpr)
inModule = is.finite(match(moduleColors, modules));
modProbes = probes[inModule];
#modGenes = annot$gene_symbol[match(modProbes, annot$substanceBXH)];
# Select the corresponding Topological Overlap
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
# Export the network into edge and node list files Cytoscape can read
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("AS-green-FPKM-Step-by-step-CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
nodeFile = paste("AS-green-FPKM-Step-by-step-CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
#altNodeNames = modGenes,
nodeAttr = moduleColors[inModule]);
#################################################################################################
#3.2.6. 分析网络可视化,用heatmap可视化权重网络,heatmap每一行或列对应一个基因,颜色越深表示有较高的邻近
#################################################################################################
options(stringsAsFactors = FALSE);
lnames = load(file = "AS-green-FPKM-01-dataInput.RData");
lnames
lnames = load(file = "AS-green-FPKM-02-networkConstruction-stepByStep.RData");
lnames
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
#====================================
#3.2.6.1. 可视化全部基因网络 ==========
#====================================
# Calculate topological overlap anew: this could be done more efficiently by saving the TOM
# calculated during module detection, but let us do it again here.
dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6);
# Transform dissTOM with a power to make moderately strong connections more visible in the heatmap
plotTOM = dissTOM^7;
# Set diagonal to NA for a nicer plot
diag(plotTOM) = NA;
# Call the plot function
#sizeGrWindow(9,9)
TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")
#====================================
#3.2.6.2. 随便选取1000个基因来可视化 ==
#====================================
nSelect = 1000
# For reproducibility, we set the random seed
set.seed(10);
select = sample(nGenes, size = nSelect);
selectTOM = dissTOM[select, select];
# There's no simple way of restricting a clustering tree to a subset of genes, so we must re-cluster.
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select];
# Open a graphical window
#sizeGrWindow(9,9)
# Taking the dissimilarity to a power, say 10, makes the plot more informative by effectively changing
# the color palette; setting the diagonal to NA also improves the clarity of the plot
plotDiss = selectTOM^7;
diag(plotDiss) = NA;
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes")
#此处画的是根据基因间表达量进行聚类所得到的各模块间的相关性图
MEs = moduleEigengenes(datExpr, moduleColors)$eigengenes
MET = orderMEs(MEs)
sizeGrWindow(7, 6)
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(3,4,2,2), plotDendrograms = FALSE, xLabelsAngle = 90)
部分结果图简单解释
Cytoscape生成网络图
只需要第二个edges文件就能构建网络图。导入该文件后,在软件的导入设置中,将第一列设置为fromNode,第二列设置为toNode,最后把第三列设为网络关系属性,完成设置,便可生成网络图了。
WGCNA样本要求
由于WGCNA是基于相关系数的表达调控网络分析方法。当样本数过低的时候,相关系数的计算是不可靠的,得到的调控网络价值不大。所以,我们推荐的样本数如下:
报错暨解决办法
错误1
运行 pickSoftThreshold 函数报错如下1
2
3
4
5> sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
pickSoftThreshold: will use block size 773.
pickSoftThreshold: calculating connectivity for given powers...
..working on genes 1 through 773 of 57862
Error in { : task 1 failed - "'x' has a zero dimension."
检查 datExpr 变量1
2> dim(datExpr)
[1] 0 50057
重新dim检查之前的datExpr变量,绝对不是0行,而 datExpr变量的最近一次处理是 keepSamples = (clust==1)
察看keepSamples = (clust==1)之后的keepSamples变量1
2> keepSamples
[1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
都是FALSE???,所以引起后续datExpr = datExpr0[keepSamples, ]的错误;
继续往上查找变量clust,即clust = cutreeStatic(sampleTree, cutHeight = 80000, minSize = 10),cutreeStatic 函数会通过cutHeight 和 minSize 的设定对聚类树进行cut去掉异常的样本,
cutHeight :height at which branches are to be cut.
minSize :minimum number of object on a branch to be considered a cluster.
所以调整相应的cutHeight 和 minSize 参数,察看 clust变量的值,保留可行的样本;1
2
3
4
5
6
7
8
9
10> clust
[1] 1 2 1 2 1 1 2 1 1 1 1 1
> keepSamples = (clust==1)
> keepSamples
[1] TRUE FALSE TRUE FALSE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE
> clust2
[1] 0 0 0 0 0 0 0 0 0 0 0 0
> keepSamples = (clust2==0)
> keepSamples
[1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
或者简单暴力手动去除刚开始输入数据中异常样本,不做 聚类检测输入样本是否含有异常值(obvious outliers)这一过程;
错误2
按照WGCNA手册第五步Network visualization using WGCNA functions时报错如下:1
2
3
4> TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")
Error in .heatmap(as.matrix(dissim), Rowv = as.dendrogram(dendro, hang = 0.1), :
row dendrogram ordering gave index of wrong length
看到row dendrogram ordering gave index of wrong length这句报错内容,分别察看plotTOM, geneTree, moduleColors这三个变量length;1
2
3> dim(plotTOM)
> geneTree
> moduleColors
果然,三者的length不同,发现geneTree少了一些,往回找geneTree来源 geneTree = net$dendrograms[[1]],net来源于网络构建过程:1
2
3
4
5
6
7net = blockwiseModules(datExpr, power = 6,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
所以,这是问题所在,继续察看文档发现基于内存等因素blockwiseModules函数默认最大maxBlockSize=5000(即最大5000个基因数目),而我们的数据超过了这个值,所以函数自动将输入datExpr数据做了拆分处理,而解决办法也很简单,设置maxBlockSize参数大于我们的值(dim(datExpr)所显示的数据行数) 即可。
相关讨论
signed vs unsigned network
I think the “sign” represents the sign of weight on the edges. If you care about the sign, for example sign represents positive or negative regulation between two nodes, then you need to choose “signed”. On the other hand, if the weight just represent the strength of relatedness between two nodes, you might don’t care positive or negative, so you need to choose “unsigned”. That really depends on your needs. 【WGCNA signed vs unsigned network】
Should you use a signed or unsigned network? By and large, I recommend using one of the signed varieties, for two main reasons. First, more often than not, direction does matter: it is important to know where node profiles go up and where they go down, and mixing negatively correlated nodes together necessarily mixes the two directions together. Second, negatively correlated nodes often belong to different categories. For example, in gene expression data, negatively correlated genes tend to come from biologically very different categories. It is true that some pathways or processes involve pairs of genes that are negatively correlated; if there are enough negatively correlated genes, they will form a module on their own and the two modules can then be analyzed together. (For the advanced practitioner, another option is to use the fuzzy module membership measure based on the module eigengene to attach a few strongly negatively correlated genes to a module after the modules have been identified). 【Signed or unsigned: which network type is preferable?】
Should I filter probesets or genes
Probesets or genes may be filtered by mean expression or variance (or their robust analogs such as median and median absolute deviation, MAD) since low-expressed or non-varying genes usually represent noise. Whether it is better to filter by mean expression or variance is a matter of debate; both have advantages and disadvantages, but more importantly, they tend to filter out similar sets of genes since mean and variance are usually related.
We do not recommend filtering genes by differential expression. WGCNA is designed to be an unsupervised analysis method that clusters genes based on their expression profiles. Filtering genes by differential expression will lead to a set of correlated genes that will essentially form a single (or a few highly correlated) modules. It also completely invalidates the scale-free topology assumption, so choosing soft thresholding power by scale-free topology fit will fail. 【Data analysis questions】
power must be between 1 and 30
First, the user should ensure that variables (probesets, genes etc.) have not been filtered by differential expression with respect to a sample trait. See item 2 above for details about beneficial and detrimental filtering genes or probesets.
If the scale-free topology fit index fails to reach values above 0.8 for reasonable powers (less than 15 for unsigned or signed hybrid networks, and less than 30 for signed networks) and the mean connectivity remains relatively high (in the hundreds or above), chances are that the data exhibit a strong driver that makes a subset of the samples globally different from the rest. The difference causes high correlation among large groups of genes which invalidates the assumption of the scale-free topology approximation.
Lack of scale-free topology fit by itself does not invalidate the data, but should be looked into carefully. It always helps to plot the sample clustering tree and any technical or biological sample information below it as in Figure 2 of Tutorial I, section 1; strong clusters in the clustering tree indicate globally different groups of samples. It could be the result a technical effect such as a batch effect, biological heterogeneity (e.g., a data set consisting of samples from 2 different tissues), or strong changes between conditions (say in a time series). One should investigate carefully whether there is sample heterogeneity, what drives the heterogeneity, and whether the data should be adjusted (see previous point).
If the lack of scale-free topology fit turns out to be caused by an interesting biological variable that one does not want to remove (i.e., adjust the data for), the appropriate soft-thresholding power can be chosen based on the number of samples as in the table below. This table has been updated in December 2017 to make the resulting networks conservative.
| Number of samples | Unsigned and signed hybrid networks | Signed networks |
|---|---|---|
| Less than 20 | 9 | 18 |
| 20-30 | 8 | 16 |
| 30-40 | 7 | 14 |
| more than 40 | 6 | 12 |
