测序技术的发展普及极快的促进了基因组学的研究进展,如果说DNA sequencing at 40: past, present and future呈现给我们的是气势磅礴的DNA测序技术,那“拟南芥1001基因组计划”(http://1001genomes.org )就是这雄伟蓝图中的冰山一角,自2008年启动伊始,这一角却撬动了生命科学研究的步伐,加速了人类对基因功能,物种多样性的认知。2016年1135 个拟南芥基因组全基因组序列分析论文的发表,宣告了 1001 基因组计划项目第一阶段的结束,然而基于基因组的拟南芥相关研究却从未止步。
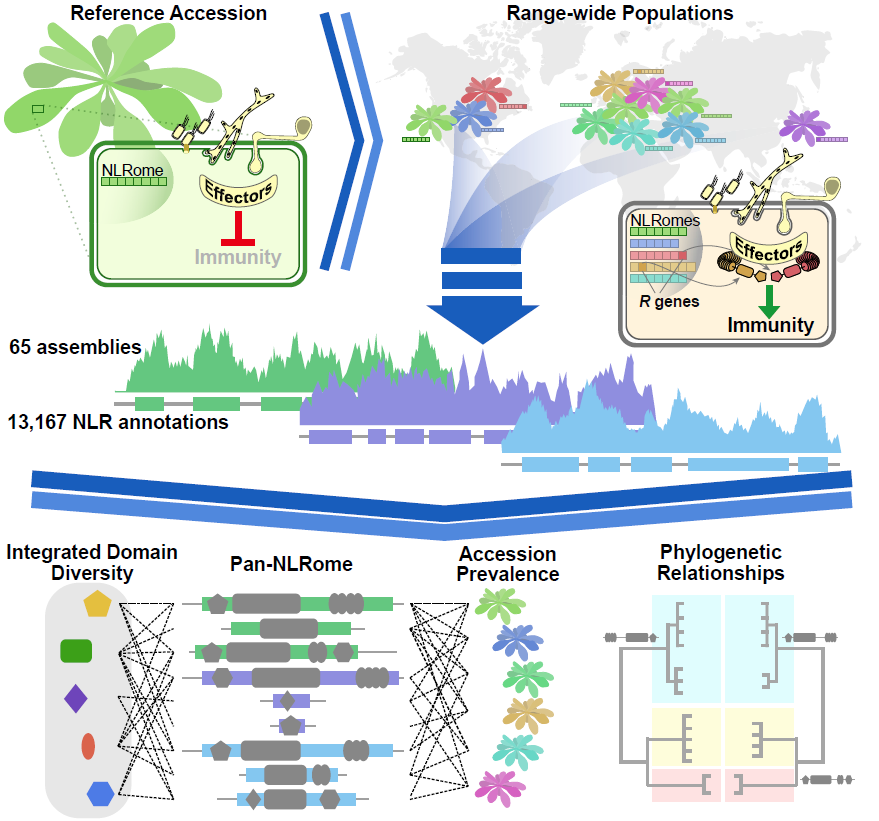
bioRxiv最近上传了题为“Chromosome-level assemblies of multiple Arabidopsis thaliana accessions reveal hotspots of genomic rearrangements”的文章通过对7个拟南芥品种进行PacBio测序和染色体水平基因组组装,揭示了其在长期进化过程中相应逆境胁迫而引起的大约350个热点区域的染色体重排。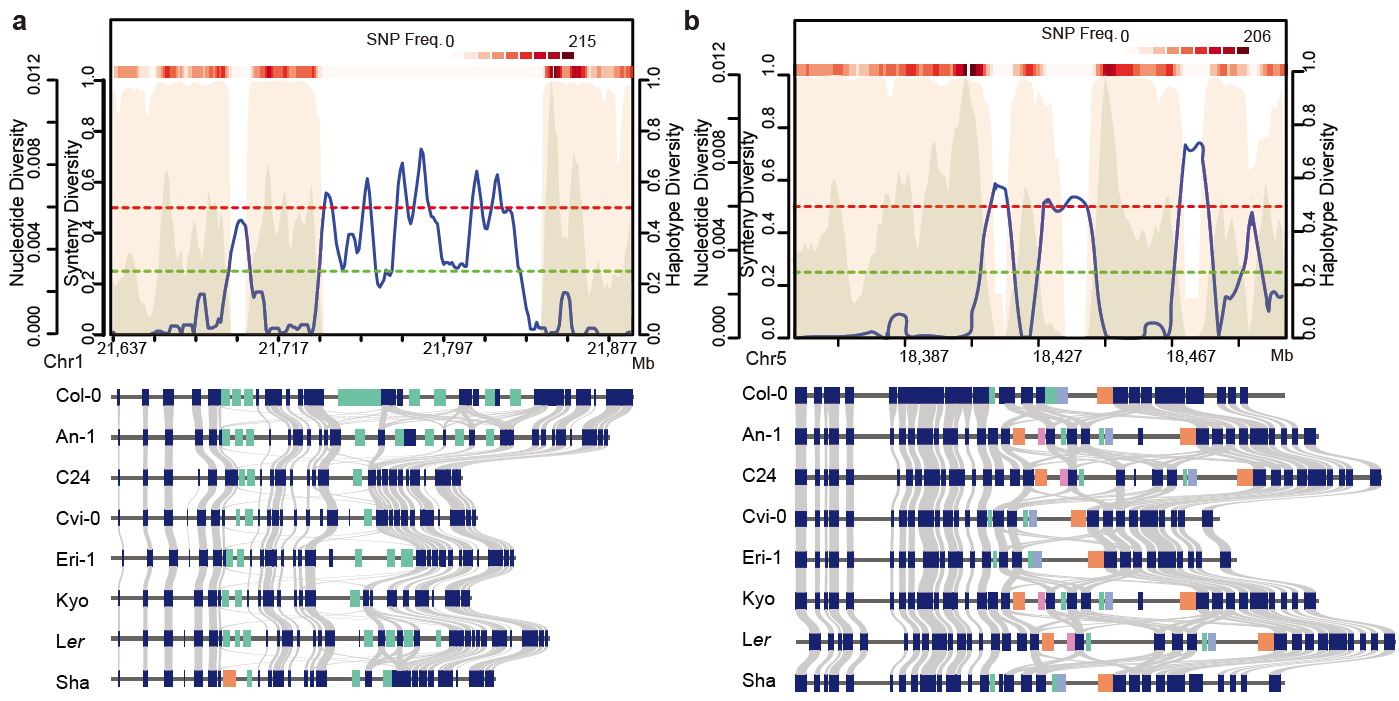
背景介绍
说起逆境胁迫,植物从寒武纪生命大爆发选择陆地作为落脚点开始,风雨不动安如山便是对它们最真实的写照,为生存植物在长期的进化过程中形成其独特的免疫系统来发现潜在的病原体,植物免疫依赖于一系列免疫受体,而植物与微生物的共进化促进了免疫受体的多样性。通常有两种类型的受体可以激活植物的免疫信号:能够识别病原相关分子模式(microbe-associated molecular patterns, MAMPs)的细胞表面蛋白和能够感知病原菌效应子的细胞内蛋白。病原菌可以通过释放一系列的效应子来破坏或扰乱植物的PTI防御反应以便更好地侵染植物,植物在PTI的基础上配置了多种类型的核苷酸结合富亮氨酸重复的免疫受体 (nucleotide-binding leucine-rich repeat, NLR),作为效应子触发免疫的第2道防御体系 (即ETI)。植物通过NLR免疫受体识别病原体,引起植物的超敏反应(hypersensitive response, HR),进而阻止病原菌的侵染。NLR 是具有保守结构的多结构域蛋白,包括C 末端富含亮氨酸重复序列(LRR)结构域,中央核苷酸结合结构域(NBD)和直接或间接识别病原体衍生效应子的可变N-末端结构域,其主要是TIR(Toll/interleukin-1 receptor)和CC(coiled-coil)结构域。就在昨天Science背靠背报道了TIR结构域能够分裂代谢辅因子NAD+(nicotinamide adenine dinucleotide),并作为抗病反应的细胞死亡信号。(详见:Science背靠背 | NLR受体介导植物抗病反应新机制)。这些关键性结构域为NLRs相关基因的鉴定和家族分析提供了依据。
基于NLR基因在植物免疫和育种应用中的重要性,目前有大量的物种进行相关基因的鉴定和进化分析。然而NLR基因家族极端的多态性,甚至是近缘个体间广泛存在的获得与缺失变异(presence-absence variants,PAVs),使得人们对NLR的多样性至今无清晰了解。
近日,来自德国马普发育生物学研究所的Felix Bemm团队在Cell杂志在线发表了“A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis thaliana” 的研究论文,通过对64个不同地理分布的拟南芥accession进行RenSeq测序(Resistance gene enrichment Sequencing,能够从已测序的植物基因组中重新定位NLR基因家族,并快速绘制分离群体中的抗性位点)和泛NLR组(pan-NLRome)分析,进而确定核心NLR complement,整合结构域多样性,描述新的结构域特征,评估非核心NLRs的获得与缺失多态性和锚定拟南芥Col-0参考基因组上的非典型NLRs基因位置。
结果
NLR Discovery
通过RenSeq和单分子实时测序(SMRT),在64个拟南芥中共构建了65个NLR complement,其中包含13,167个注释的NLR基因,平均在每个个体中有167到251个基因。其中47%到71%的NLR基因在不同个体基因组中成簇存在,且部分NLR基因呈现出head-to-head的方向性(定义为paired NLRs),每个accession中有10到34个这个的配对NLRs。所有NLRs基因根据其所包含结构域的不同分为四类:TIR-NLR (TNL), CC-NLR (CNL), CCR-NLR (RNL), 和NB-and-LRR-only proteins (NL),其中每个accession中数量最多的是TNLs,其次是NLs, CNLs和RNLs。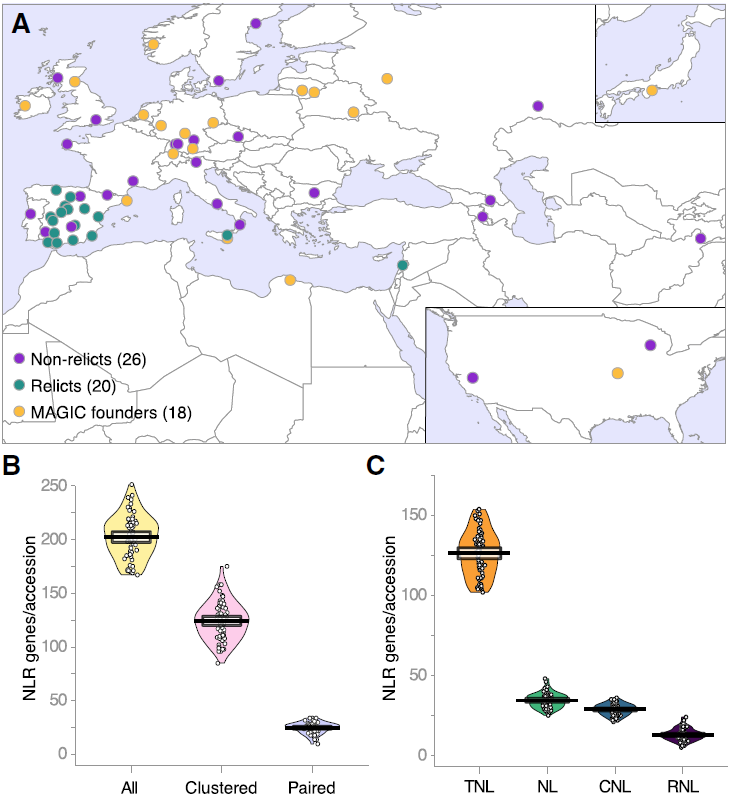
Diversity of NLR Domain Architectures
这13,167个NLR基因中,663个编码至少一个非典型NLR结构域,代表36个明显不同的Pfam结构域。NLR组跨物种多样性的显著标志是不同结构域特征相对比例的变化。本次拟南芥泛NLR组研究共鉴定到97个明显的特征,其中仅有22个在Col-0参考基因组中存在,仅有48个在Col-0或其他十字花科植物中报道过。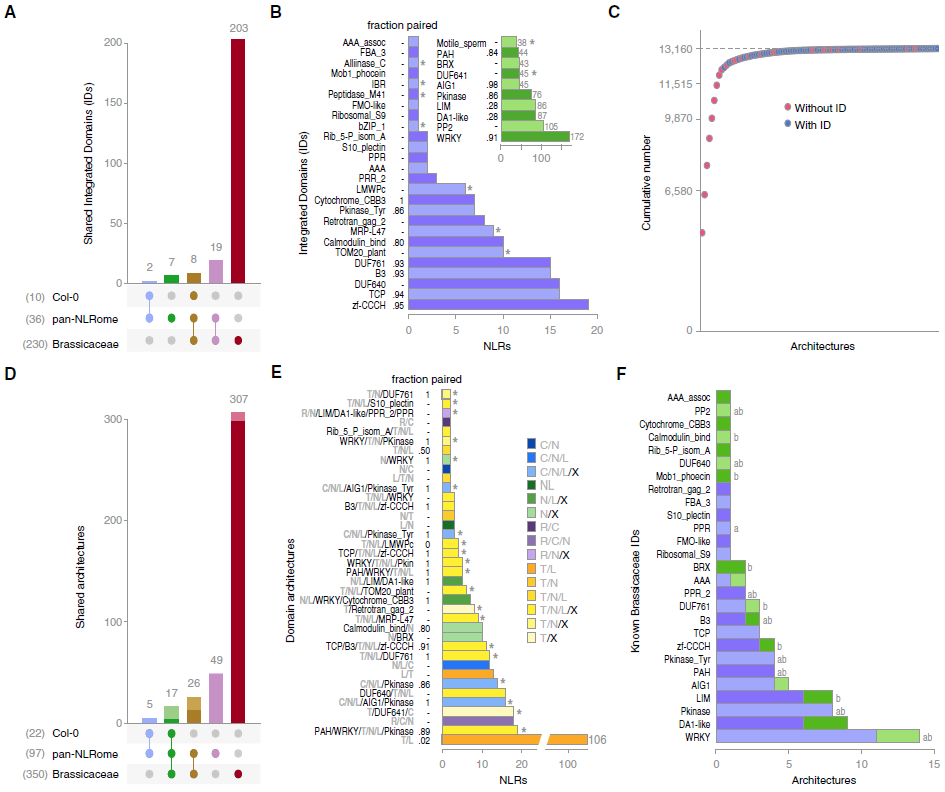
The Pan-NLRome
为了解NLR数量和多样性变异,对所测序的64个拟南芥accessions所含NLRs基因基于序列相似性进行orthogroups(OGs)聚类。结果仅有小于10%(1,663个基因)的基因以单体形式存在,剩下的11,497个基因共聚成464个OGs。这464个OGs中95%可在任意的38个accessions中找到。OGs进一步通过大小,结构域特征和结构特征进行分类。核心NLR组仅包含106个OGs(23%),对应6,080(53%)个基因能在至少52个accessions中找到。shell NLR组有稍微高的143个OGs (31%),对应3,932 (34%)个基因能在至少13个,但少于52个accessions中找到。cloud NLR组为215个OGs (46%),对应1,485 (13%)个基因能在最多12个accessions中找到。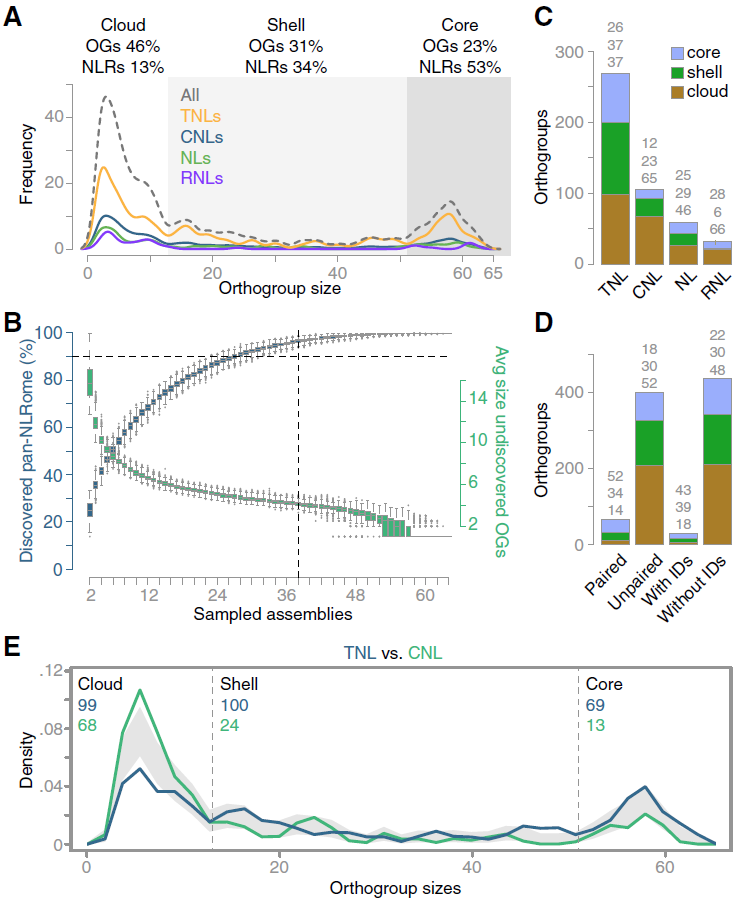
Genomic Placement of Non-reference OGs
296个OGs在Col-0参考基因组中缺失,其中6个属于核心,205个属于cloud和85个属于shell NLR组。本研究通过共线性将OGs锚定到Col-0参考基因组。结果共得到42个共线性子网络。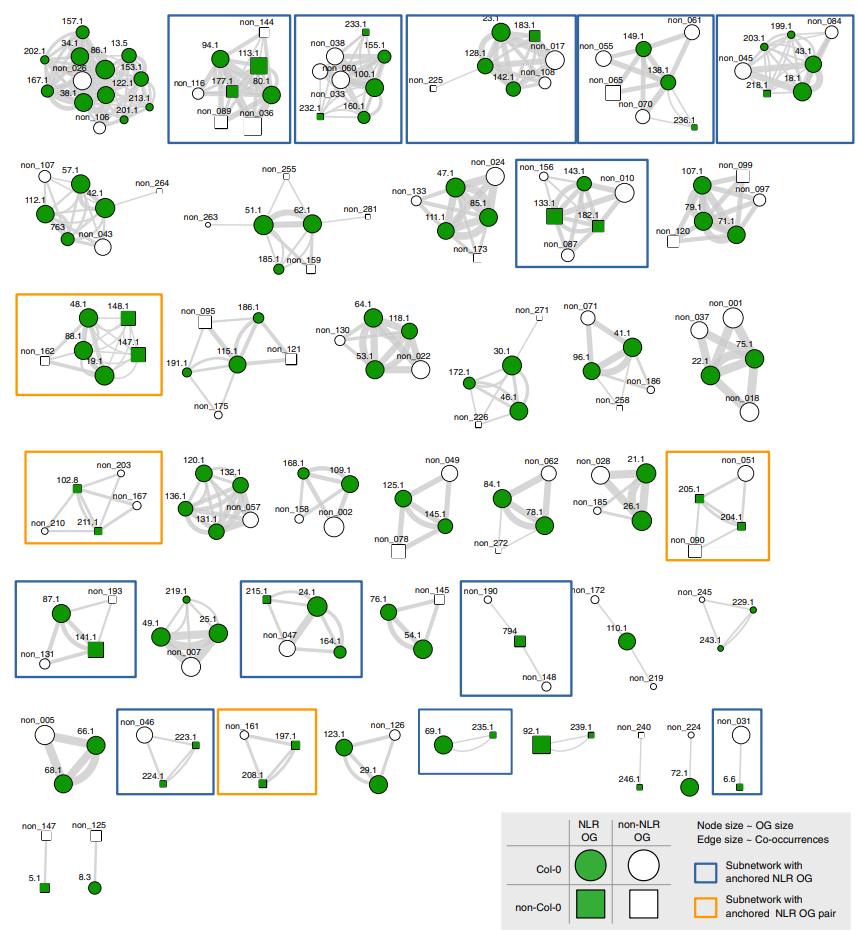
其中OG102和OG211聚类在新的NLRs区域中,此外,新锚定的OGs还包括一个CNL和三个TNL类的 NLR基因对。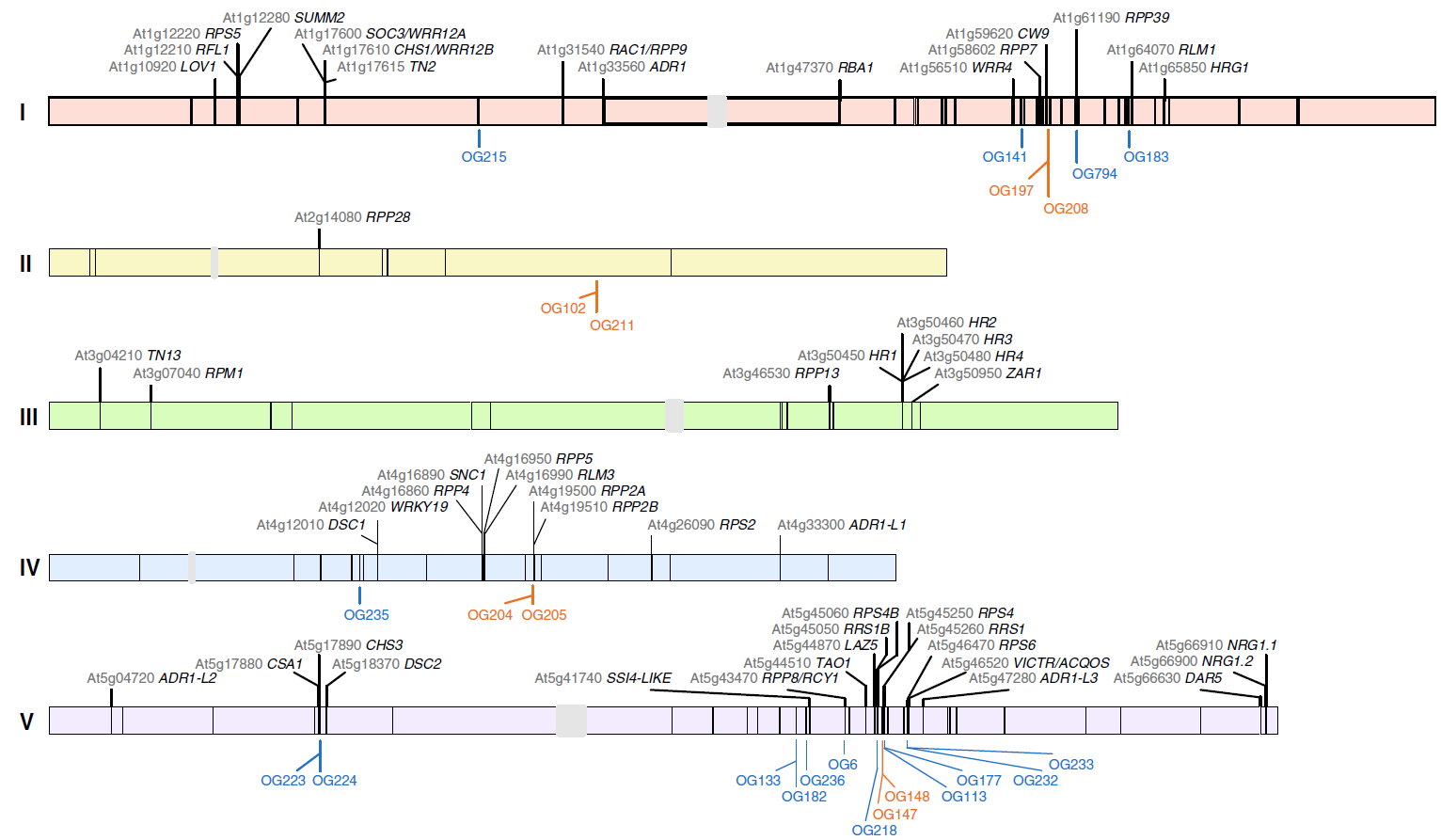
Pan-NLRome Diversity
基于正交分类法(orthogonal approach, http://www.pharmtech.com/orthogonal-approach-biosimilarity ) 根据结构域特征对NLR基因进行分类,并评估了序列多样性作为形成泛NLR组的进化压的指示。随机挑选的32个accessions中,平均核苷酸序列多样性达到95%的饱和度。相反,仅有49个accessions存在单倍型多样性的饱和度,这反映出新的单倍型的出现不仅来源于突变,而且与基因内的重排和基因转换相关,这在3/4的OGs(74%)中得到验证。这与基因内的重排能引起功能分化的报道相吻合。相较于没有聚类的OGs而言,成簇的OGs有显著高的核苷酸多样性,这与基因复制后存在较小的选择压相符合。尽管当涉及到获得与缺失多态性时不同的NLR分类有不同的整体轮廓,但是平均核苷酸多样性在OGs内是相似的对于CNLs, TNLs和NLs类NLR组来说。而RNLs类有较低的平均核苷酸多样性,这与他们功能的保守性相关联。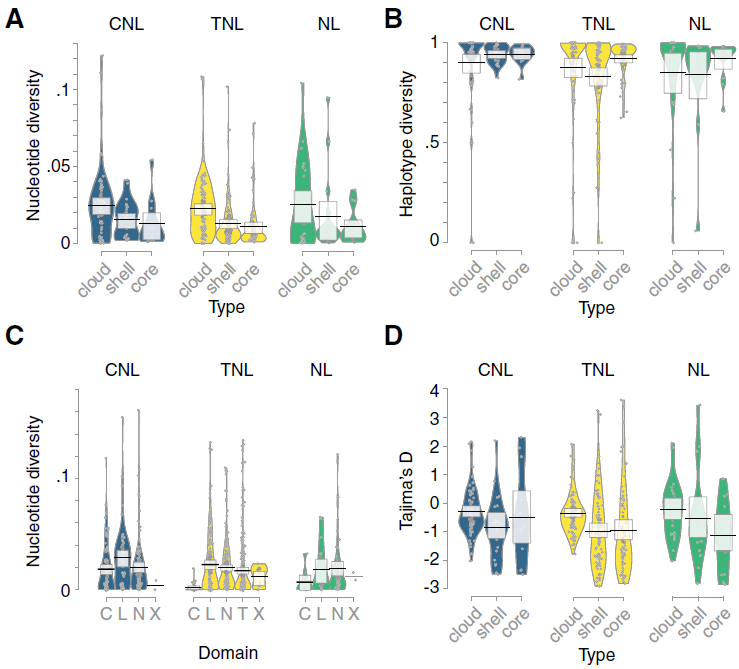
对进化的每一个分支进行选择分析共鉴定到131个OGs在至少一个分支中存在偶然正选择(episodic positive selection),大部分OGs属于核心(50)或shell (73)NLR组。位点特异的选择分析揭示了543个核心和shell OGs可能经历过experienced constant (46%), pervasive (30%)或 episodic (24%)正选择。没有变化的密码子代表了恒定的纯化选择能够在所有类型(core和shell),分类(TNLs, CNLs,RNL和NL)和配对状态 (paired和unpaired)中找到,而其子类表现出不均匀的正选择模式。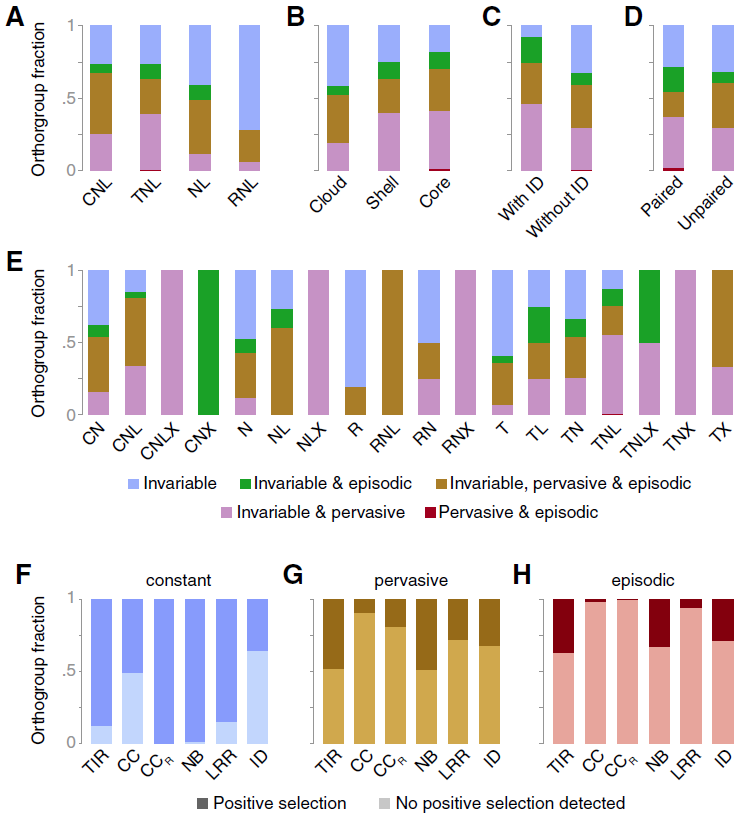
Linking Diversity to Known Function
为了将NLR组多样性与已知的基因功能相联系,进一步将OGs分类为对适应性活体营养型有抗性,对非适应性活体营养型有抗性和对半活体营养型有抗性三类。结果显示OGs中对适应性活体营养型(adapted biotrophs)有抗性类型显著多于其他类别,说明了宿主适应性活体营养型病原菌更加能够驱动NLRs基因的多样性。对适应性活体营养型有抗性的OGs有较高的Tajima’s D值,表明其不仅经历过正向选择也经历过平衡选择。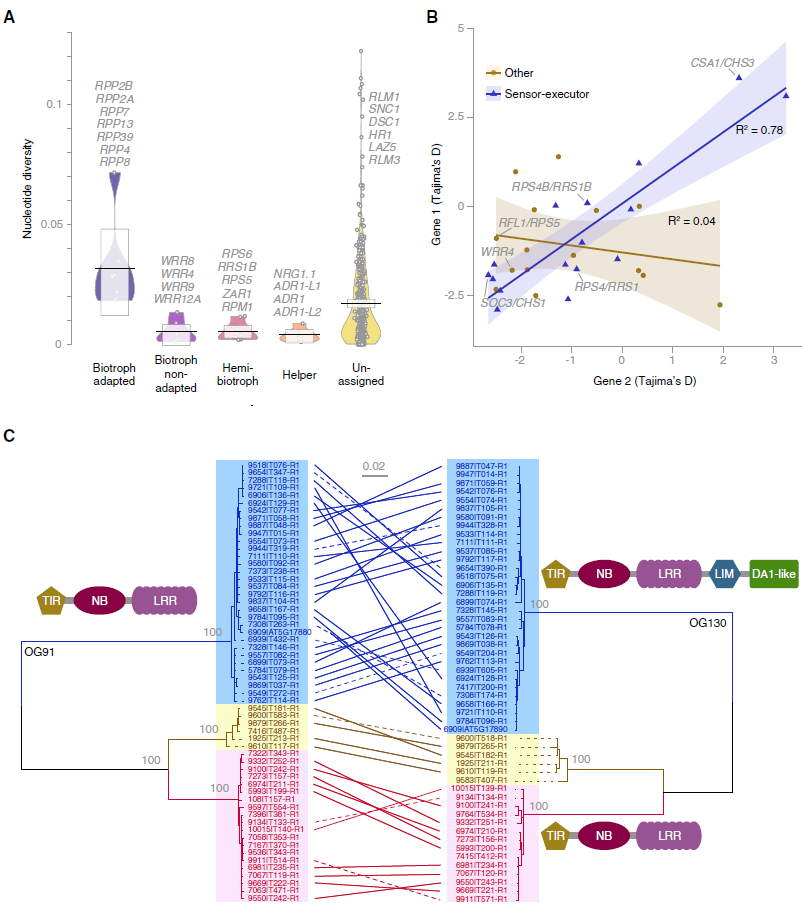
数据分析
正文结束,我是分割线
纵观全文,这个基因家族分析涉及SMRT RenSeq测序,类似于基因组的De novo组装,基因结构和功能注释以及分类,其中为保证NLR基因注释准确性通过多种手段进行人工纠正,随后的比较基因组分析,泛NLR组分析,聚类分析和序列水平的选择分析,最关键的是每一个漂亮的figure都告诉你是怎么画的,包括R脚本。正所谓面面俱到,完全不像满大街的单个物种找下目标基因,看下染色体分布,什么基因结构,motif有用没用先放上,或者再计算个简单的kaks,最后东拼西凑点转录组数据,塞几个qRT结果……类基因家族分析文章。非常值得学习。
最后奉上师兄的点评。
原文链接: A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis thaliana
