This tutorial describes how to create a graph with error bars using R software and ggplot2 package. There are different types of error bars which can be created using the functions below :
- geom_errorbar()
- geom_linerange()
- geom_pointrange()
- geom_crossbar()
- geom_errorbarh()
Add error bars to a bar and line plots
Prepare the data
ToothGrowth data is used. It describes the effect of Vitamin C on tooth growth in Guinea pigs. Three dose levels of Vitamin C (0.5, 1, and 2 mg) with each of two delivery methods [orange juice (OJ) or ascorbic acid (VC)] are used :
1 | library(ggplot2) |
In the example below, we’ll plot the mean value of Tooth length in each group. The standard deviation is used to draw the error bars on the graph.
First, the helper function below will be used to calculate the mean and the standard deviation, for the variable of interest, in each group :
1 | #+++++++++++++++++++++++++ |
Summarize the data :
1 | df2 <- data_summary(ToothGrowth, varname="len", |
Barplot with error bars
The function geom_errorbar() can be used to produce the error bars :
1 | library(ggplot2) |
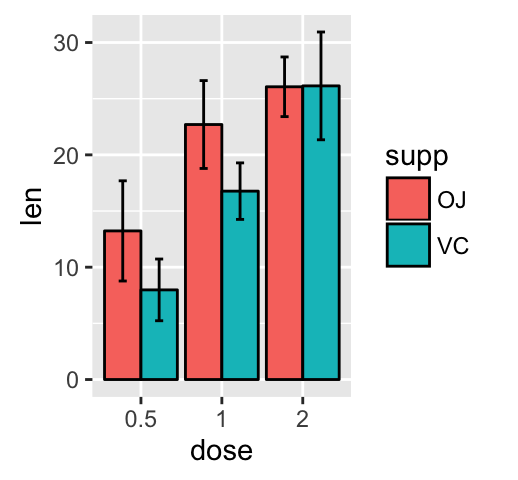
Note that, you can chose to keep only the upper error bars
1 | # Keep only upper error bars |
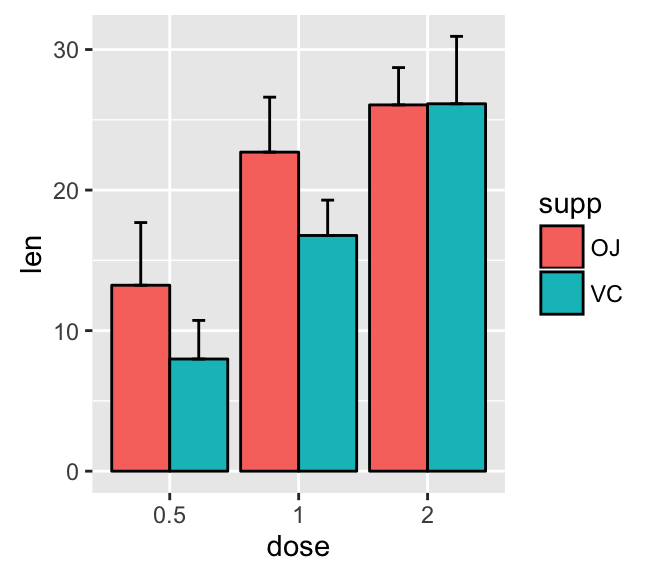
Read more on ggplot2 bar graphs : ggplot2 bar graphs
Line plot with error bars
1 | # Default line plot |
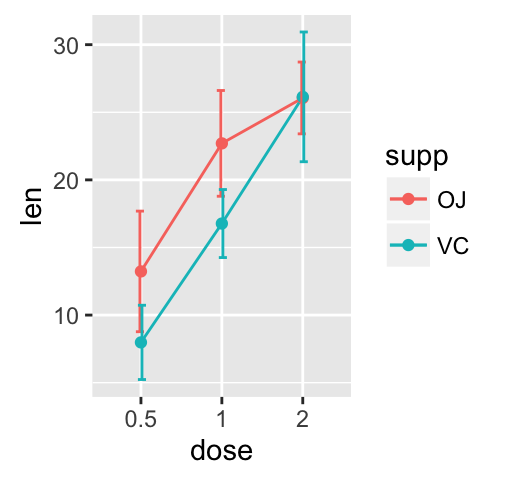

You can also use the functions geom_pointrange() or geom_linerange() instead of using geom_errorbar()
1 | # Use geom_pointrange |


Read more on ggplot2 line plots : ggplot2 line plots
Dot plot with mean point and error bars
The functions geom_dotplot() and stat_summary() are used :
The mean +/- SD can be added as a crossbar , a error bar or a pointrange :
1 | p <- ggplot(df, aes(x=dose, y=len)) + |
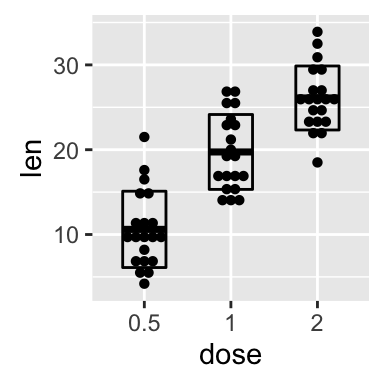
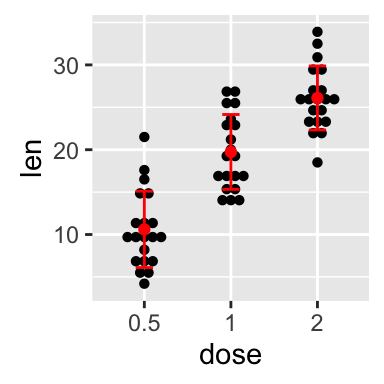
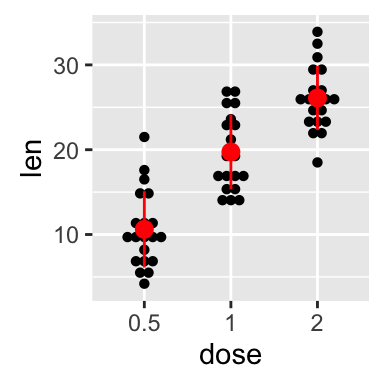
Read more on ggplot2 dot plots : ggplot2 dot plot
Infos
This analysis has been performed using R software (ver. 3.1.2) and ggplot2 (ver. 1.0.0)
Contribution from :http://www.sthda.com/english/wiki/ggplot2-error-bars-quick-start-guide-r-software-and-data-visualization