概述:所有的类linux系统都会提供一个名为grep(global regular expression print,全局正则表达式输出)的搜索工具。grep命令在对一个或多个文件的内容进行基于模式的搜索的情况下是非常有用的。模式可以是单个字符、多个字符、单个单词、或者是一个句子。
当命令匹配到执行命令时指定的模式时,grep会将包含模式的一行输出,但是并不对原文件内容进行修改。
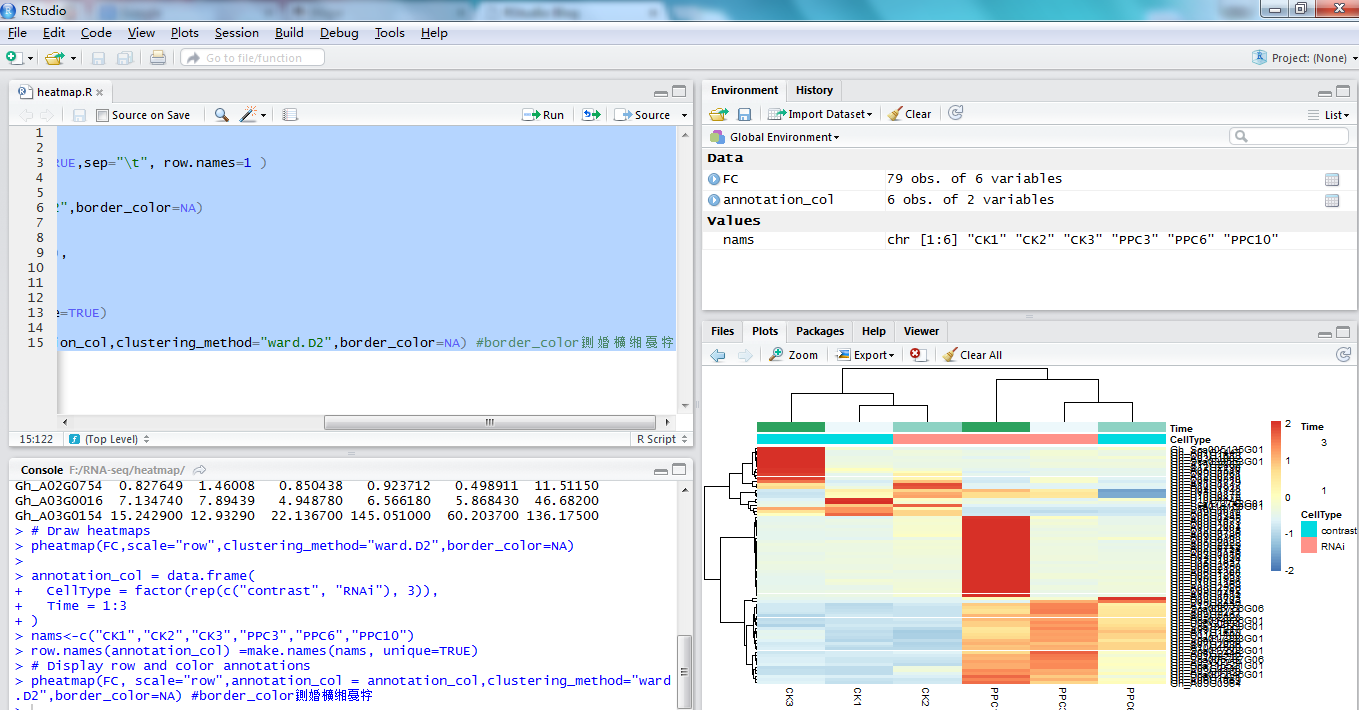
在本文中,我们将会讨论到14个grep命令的例子。
例1 在文件中查找模式(单词)在/etc/passwd文件中查找单词”linuxtechi”
1 | root@Linux-world:~# grep linuxtechi /etc/passwd |
例2 在多个文件中查找模式。
1. root@Linux-world:~# grep linuxtechi /etc/passwd /etc/shadow /etc/gshadow
2. /etc/passwd:linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
3. /etc/shadow:linuxtechi:$6$DdgXjxlM$4flz4JRvefvKp0DG6re:16550:0:99999:7:::/etc/gshadow:adm:*::syslog,linuxtechi
4. /etc/gshadow:cdrom:*::linuxtechi
5. /etc/gshadow:sudo:*::linuxtechi
6. /etc/gshadow:dip:*::linuxtechi
7. /etc/gshadow:plugdev:*::linuxtechi
8. /etc/gshadow:lpadmin:!::linuxtechi
9. /etc/gshadow:linuxtechi:!::
10. /etc/gshadow:sambashare:!::linuxtechi
11. root@Linux-world:~#
例3 使用-l参数列出包含指定模式的文件的文件名。
1. root@Linux-world:~# grep -l linuxtechi /etc/passwd /etc/shadow /etc/fstab /etc/mtab
2. /etc/passwd
3. /etc/shadow
4. root@Linux-world:~#
例4 使用-n参数,在文件中查找指定模式并显示匹配行的行号
1. root@Linux-world:~# grep -n linuxtechi /etc/passwd
2. 39:linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
3. root@Linux-world:~#
4. root@Linux-world:~# grep -n root /etc/passwd /etc/shadow
例5 使用-v参数输出不包含指定模式的行输出/etc/passwd文件中所有不含单词”linuxtechi”的行
1. root@Linux-world:~# grep -v linuxtechi /etc/passwd
例6 使用 ^ 符号输出所有以某指定模式开头的行Bash脚本将 ^ 符号视作特殊字符,用于指定一行或者一个单词的开始。例如输出/etc/passes文件中所有以”root”开头的行
1. root@Linux-world:~# grep ^root /etc/passwd
2. root:x:0:0:root:/root:/bin/bash
3. root@Linux-world:~#
例7 使用 $ 符号输出所有以指定模式结尾的行。输出/etc/passwd文件中所有以”bash”结尾的行。
1. root@Linux-world:~# grep bash$ /etc/passwd
2. root:x:0:0:root:/root:/bin/bash
3. linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
4. root@Linux-world:~#
Bash脚本将美元($)符号视作特殊字符,用于指定一行或者一个单词的结尾。
例8 使用 -r 参数递归地查找特定模式
1. root@Linux-world:~# grep -r linuxtechi /etc/
2. /etc/subuid:linuxtechi:100000:65536
3. /etc/group:adm:x:4:syslog,linuxtechi
4. /etc/group:cdrom:x:24:linuxtechi
5. /etc/group:sudo:x:27:linuxtechi
6. /etc/group:dip:x:30:linuxtechi
7. /etc/group:plugdev:x:46:linuxtechi
8. /etc/group:lpadmin:x:115:linuxtechi
9. /etc/group:linuxtechi:x:1000:
10. /etc/group:sambashare:x:131:linuxtechi
11. /etc/passwd-:linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
12. /etc/passwd:linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
13. ............................................................................
上面的命令将会递归的在/etc目录中查找”linuxtechi”单词
例9 使用 grep 查找文件中所有的空行
1. root@Linux-world:~# grep ^$ /etc/shadow
2. root@Linux-world:~#
由于/etc/shadow文件中没有空行,所以没有任何输出
例10 使用 -i 参数查找模式grep命令的-i参数在查找时忽略字符的大小写。
我们来看一个例子,在paswd文件中查找”LinuxTechi”单词。
1. nextstep4it@localhost:~$ grep -i LinuxTechi /etc/passwd
2. linuxtechi:x:1001:1001::/home/linuxtechi:/bin/bash
3. nextstep4it@localhost:~$
例11 使用 -e 参数查找多个模式例如,我想在一条grep命令中查找’linuxtechi’和’root’单词,使用-e参数,我们可以查找多个模式。
1. root@Linux-world:~# grep -e "linuxtechi" -e "root" /etc/passwd
2. root:x:0:0:root:/root:/bin/bash
3. linuxtechi:x:1000:1000:linuxtechi,,,:/home/linuxtechi:/bin/bash
#或者
4. root@Linux-world:~# grep -E "(Olfr1413|Olfr1411)" Mus_musculus.GRCm38.75_chr1_genes.txt
ENSMUSG00000058904 Olfr1413
ENSMUSG00000062497 Olfr1411
#或者
grep 'usrquota\|grpquota' /etc/fstab
例12 使用 -f 用文件指定待查找的模式首先,在当前目录中创建一个搜索模式文件”grep_pattern”,我想文件中输入的如下内容。
1. root@Linux-world:~# cat grep_pattern
2. ^linuxtechi
3. root
4. false$
5. root@Linux-world:~#
现在,试试使用grep_pattern文件进行搜索
1. root@Linux-world:~# grep -f grep_pattern /etc/passwd
例13 使用 -c 参数计算模式匹配到的数量继续上面例子,我们在grep命令中使用-c命令计算匹配指定模式的数量
1. root@Linux-world:~# grep -c -f grep_pattern /etc/passwd
2. 22
3. root@Linux-world:~#
例14 输出匹配指定模式行的前或者后面N行
a)使用-B参数输出匹配行的前4行
1. root@Linux-world:~# grep -B 4 "games" /etc/passwd
b)使用-A参数输出匹配行的后4行
1. root@Linux-world:~# grep -A 4 "games" /etc/passwd
c)使用-C参数输出匹配行的前后各4行
1. root@Linux-world:~# grep -C 4 "games" /etc/passwd
例15 -E参数用扩展的正则表达式
列出当前目录下包含 s..s 的文件名,\1为反向引用()中内容,如下例子中等同于s。
ls | grep -E '(s).+\1'
例16 组分隔符
‘—group-separator=STRING’
当使用’-A’, ‘-B’ or ‘-C’时,使用STRING替代默认的组分隔符。
(注:组分隔符表示匹配到的内容的上下文。例如”-A 2”,在某行匹配到时,还将输出后两行,这是一个组。下一次匹配成功时,如果是在该组之后行匹配上的,则这两组中间默认使用”—“分隔)
‘—no-group-separator’
当使用’-A’, ‘-B’ or ‘-C’时,不输出任何组分隔符,而是将不同组相邻输出。