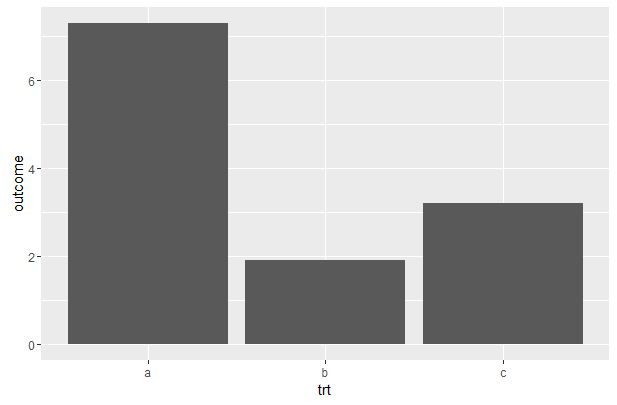
Summary Statistics
mean(),max(),min(),range(),sd()
以上函数运算中,若存在NA,则返回结果为NA,设置na.rm=TRUE忽略NA值。
mean()中移除异常值:trim(mean(x, trim = 0.1):先把x的最大的10%的数和最小的10%的数去掉,然后剩下的数算平均)
range():同时返回最大/最小值。
sd():标准差。
quantile(),fivenum(),IQR()
quantile(dow30$Open, probs=c(0,0.25,0.5,0.75,1.0))
返回不同的百分位数值,probs指定百分位。
fivenum():返回(minimum, 25th percentile, median, 75th percentile, and maximum)。
IQR():返回25%与75%的差值
以上函数既可用于单独数组,也可用于apply, tapply对数据框的操作。
summary()
对于数值变量计算了五个分位点和均值,对于分类变量则计算了频数。
Statistical Tests
基于正态分布的检验;自然群体大都为正态分布。
Comparing means
Specifically, suppose that you have a set of observations
x1, x2, …, xn with experimental mean μ and want to know if the experimental
mean is different from the null hypothesis mean μ0. Furthermore, assume that the
observations are normally distributed. To test the validity of the hypothesis, you can
use a t-test. In R, you would use the function t.test;
Comparing paired data(means )
For example, you might have two observations per subject: one before an experiment and one after the experiment.
In this case, you would use a paired t-test. You can use the t.test function, specifyingpaired=TRUE, to perform this test.
Comparing variances of two populations
To compare the variances of two samples from normal populations, R includes thevar.test function which performs an F-test;
Comparing means across more than two groups
ANOVA单因素方差分析与R实现:http://tiramisutes.github.io/2015/10/08/ANOVA.html
Correlation tests
If you’d like to check whether there is a statistically significant
correlation between two vectors, you can use the cor.test function;