1. PacBio原始下机bam格式数据上传
自PacBio Sequel平台开始,PacBio原始下机数据均为bam格式,该如何上传NCBI呢?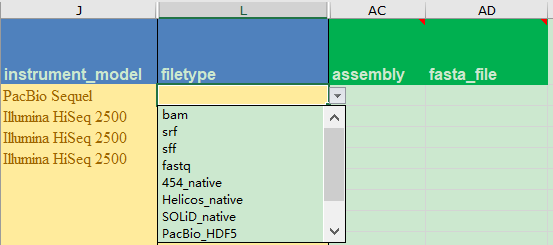
NCBI的SRA_metadata_acc.xlsx文件提供PacBio格式为PacBio RS平台的HDF5格式,而第一个bam格式则认为是比对结果文件,需要在assembly列提供比对基因组信息;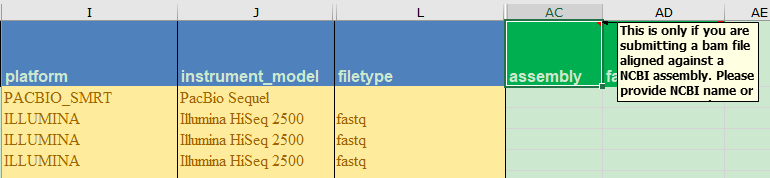
这该如何是好?万能的NCBI工作人员给我们支招了👇
For unaligned bam files please enter ‘unaligned’ in the ‘assembly’ column.
2. BioNano数据上传
进入Supplementary Files,选择BioNano原始Map数据或混合组装 (hybrid assembly) 过程数据上传;
根据介绍可选择文件类型为:CMAP,COORD (混合组装过程),XMAP,SMAP (结构变异数据) 和下机数据BNX;
3. 基因组数据上传
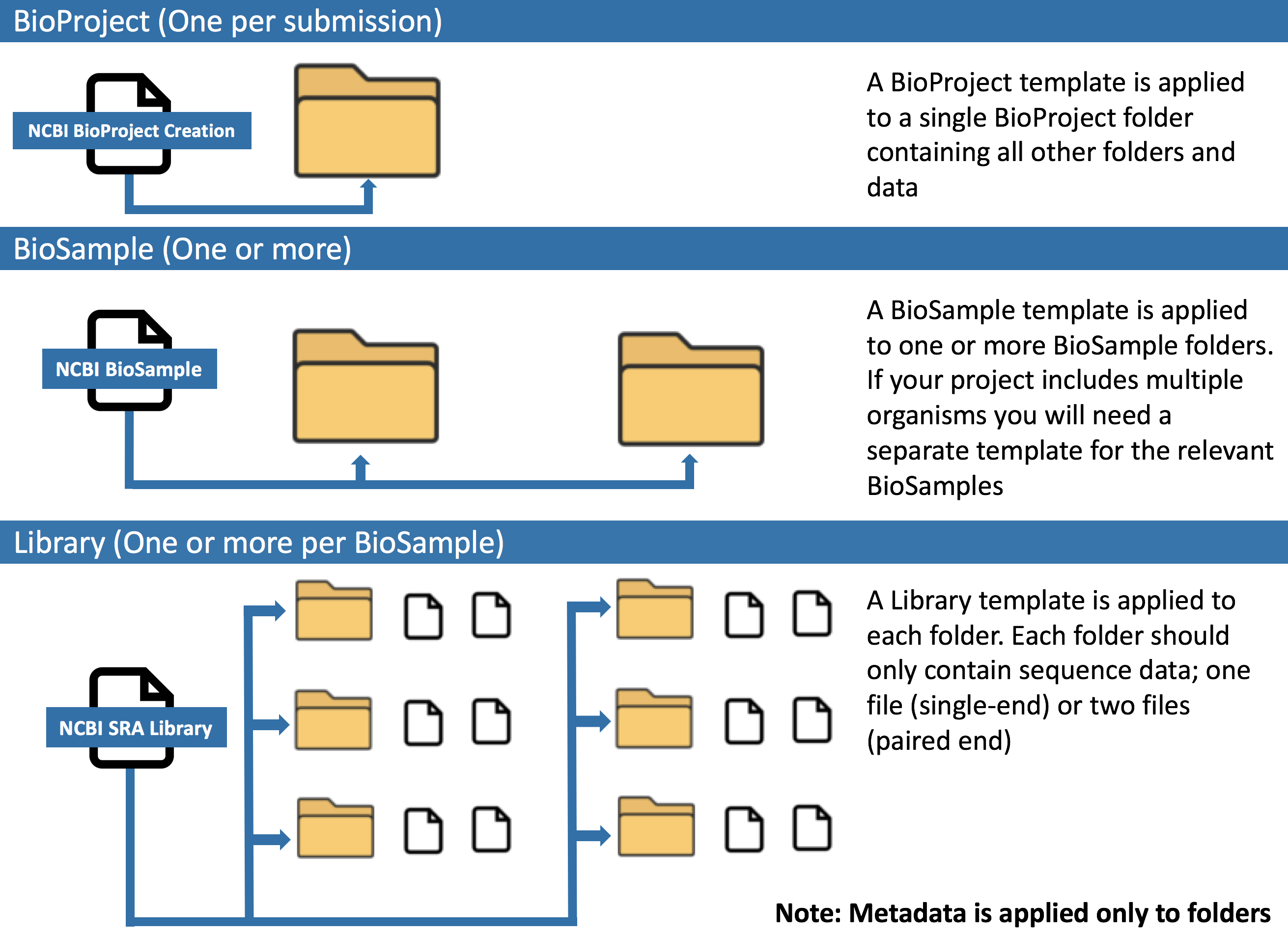
Denovo组装基因组上传时通常需上传测序相关原始数据,首先参考测序数据上传NCBI总结提交专门上传基因组测序原始数据的BioProject和BioSample;
准备基因组fa(未注释基因组)/sqn(已注释基因组)格式文件,进入Genome上传;
3.1 准备数据清单
- 基因组fa/sqn文件
- BioProject 号
- BioSample 号
- WGS 或 non-wgs genome
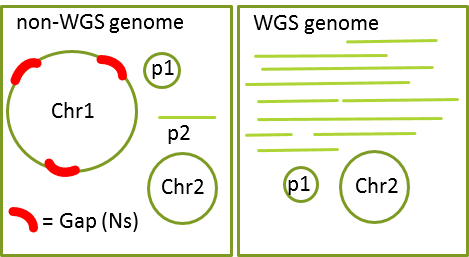
- AGP文件,可通过AGP validation on-line进行文件格式确认或者下载软件在命令行确认 (fatoagp可根据fa文件生成AGP文件; fasta2apg.pl根据fa文件生成AGP文件且输出分隔的contig.fa和scaffold.fa)
- 其他可选注释信息
4. tbl2asn 使用
tbl2asn主要用于命令行下生成 gb 或 sqn 格式文件来提交数据到GenBank数据库;
直接输入tbl2asn -来察看详细参数;
详细准备数据见: Submission using tbl2asn,即新建目录来存储相关数据,且除template.sbt外,其他文件均使用相同前缀名称;1
2
3
4template.sbt (this is the only file whose prefix is different. Leave the prefix as is).
chr01.fsa
chr01.tbl
chr01.qvl
4.1 特殊参数解释
-i指定fa文件名,且一定不能包含路径,只能是文件名;-M参数会覆盖部分其他参数;-l只有当-M设置为t时才可用;-a s: fa文件包含多个序列,结果生成单个提交文件;
4.2 运行实例
1 | cd NCBI_Genome |
4.3 流程化运行
请参考: WGS2NCBI - toolkit for preparing genomes for submission to NCBI
5. fasta+GFF 转 Genebank/EMBL
参考 Genome_Scripts 项目👇1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96#!/usr/bin/env python
"""Convert a GFF and associated FASTA file into GenBank format.
Usage:
gff_convert.py -f genbank -s <GFF annotation file> <FASTA sequence file>
"""
import sys
import os
from Bio import SeqIO
from Bio.Alphabet import generic_dna
from Bio import Seq
import argparse
from BCBio import GFF
parser=argparse.ArgumentParser(
description='''Script that converts GFF + Fasta to GBK or EMBL ''',
epilog="""hope (2019) http://tiramisutes.github.io/2019/04/05/PBGNCBI.html""")
parser.add_argument("gff", help='GFF file')
parser.add_argument("fasta", help='Fasta file')
parser.add_argument("-f", "--format", choices=['genbank', 'embl'])
parser.add_argument("-s","--split", action='store_true', help='Split output into single files, 1 per contig')
parser.add_argument("-o","--output", help='Set the directory of output file/files')
args=parser.parse_args()
if len(sys.argv) < 2:
parser.print_usage()
sys.exit(1)
def _fix_ncbi_id(fasta_iter):
"""GenBank identifiers can only be 16 characters; try to shorten NCBI.
"""
for rec in fasta_iter:
if len(rec.name) > 16 and rec.name.find("|") > 0:
new_id = [x for x in rec.name.split("|") if x][-1]
print "Warning: shortening NCBI name %s to %s" % (rec.id, new_id)
rec.id = new_id
rec.name = new_id
yield rec
def _check_gff(gff_iterator):
"""Check GFF files before feeding to SeqIO to be sure they have sequences.
"""
for rec in gff_iterator:
if isinstance(rec.seq, Seq.UnknownSeq):
print "Warning: FASTA sequence not found for '%s' in GFF file" % (
rec.id)
rec.seq.alphabet = generic_dna
yield _flatten_features(rec)
def _flatten_features(rec):
"""Make sub_features in an input rec flat for output.
GenBank does not handle nested features, so we want to make
everything top level.
"""
out = []
for f in rec.features:
cur = [f]
while len(cur) > 0:
nextf = []
for curf in cur:
out.append(curf)
if len(curf.sub_features) > 0:
nextf.extend(curf.sub_features)
cur = nextf
rec.features = out
return rec
gff_file = args.gff
fasta_file = args.fasta
format = args.format
output_dir = args.output
if args.split:
if format == "genbank":
print("Output set to " + format + ", splitting files and writting individual records to directory: " + output_dir)
fasta_input = SeqIO.to_dict(SeqIO.parse(fasta_file, "fasta", generic_dna))
for rec in GFF.parse(gff_file, fasta_input):
SeqIO.write(_check_gff(_fix_ncbi_id([rec])), open(output_dir + "/" + rec.id + ".gbk", "w"), "genbank")
if format == "embl":
print("Output set to " + format + ", splitting files and writting individual records to directory: " + output_dir)
fasta_input = SeqIO.to_dict(SeqIO.parse(fasta_file, "fasta", generic_dna))
for rec in GFF.parse(gff_file, fasta_input):
SeqIO.write(_check_gff(_fix_ncbi_id([rec])), open(output_dir + "/" + rec.id + ".embl", "w"), "embl")
else:
if format == "genbank":
out_file = output_dir + "/%s.gb" % os.path.splitext(os.path.basename(gff_file))[0]
print("Output set to " + format + ", writing file to " + out_file)
fasta_input = SeqIO.to_dict(SeqIO.parse(fasta_file, "fasta", generic_dna))
gff_iter = GFF.parse(gff_file, fasta_input)
SeqIO.write(_check_gff(_fix_ncbi_id(gff_iter)), out_file, "genbank")
if format == "embl":
out_file = output_dir + "/%s.embl" % os.path.splitext(os.path.basename(gff_file))[0]
print("Output set to " + format + ", writing file to " + out_file)
fasta_input = SeqIO.to_dict(SeqIO.parse(fasta_file, "fasta", generic_dna))
gff_iter = GFF.parse(gff_file, fasta_input)
SeqIO.write(_check_gff(_fix_ncbi_id(gff_iter)), out_file, "embl")
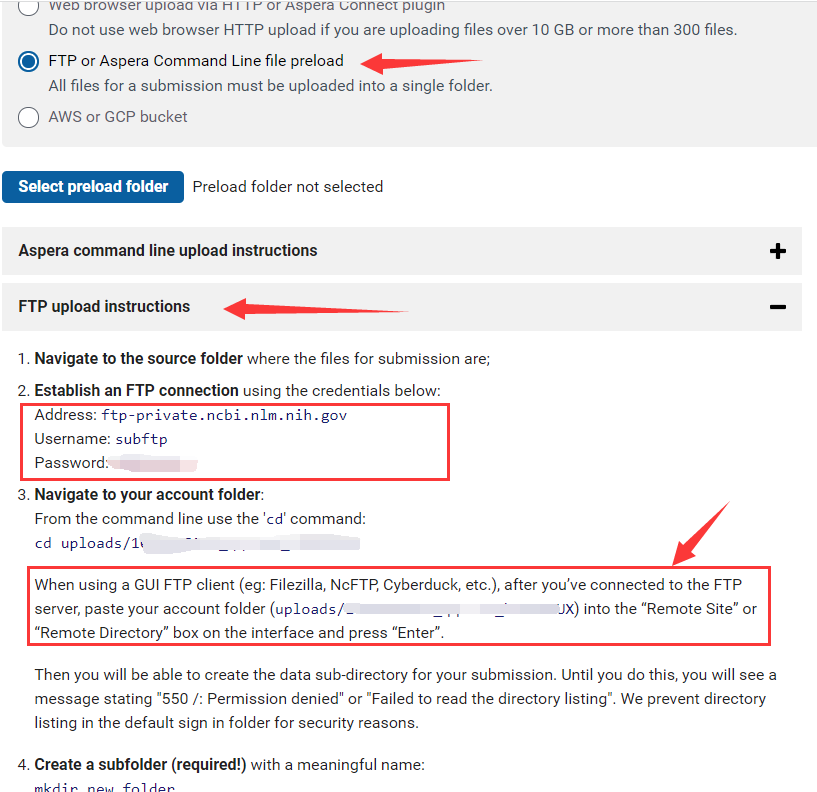
6. 通过FileZilla上传
完成SRA前面内容填充后即可到如下页面,记得使用FileZilla提交时一定要设置Remote Site。