
分子生物学(Molecular biology)是对生物在分子层次上的研究,是生物学和化学之间跨学科的研究,其研究领域涵盖了遗传学、生物化学和生物物理学等学科。分子生物学主要致力于对细胞中不同系统之间相互作用的理解,包括DNA,RNA和蛋白质生物合成之间的关系以及了解它们之间的相互作用是如何被调控的。 在我们的研究中主要涉及基因功能和代谢通路解析;
生物信息学(Bioinformatics)利用应用数学、信息学、统计学和计算机科学的方法研究生物学的问题。生物信息学的研究材料和结果就是各种各样的生物学数据,其研究工具是计算机,研究方法包括对生物学数据的搜索(收集和筛选)、处理(编辑、整理、管理和显示)及利用(计算、模拟)。当前主要的研究方向有:序列比对、序列组装、基因识别、基因重组、蛋白质结构预测、基因表达、蛋白质反应的预测,以及创建进化模型。
自迈克尔·沃特曼(Michael Waterman)率先将数学和计算方法引入生物学研究开始,如今这门交叉学科作为后起之秀正逐渐渗透到生物学研究的多个领域,21世纪是生命科学的世纪,但也是一个信息化的时代,如果做分子的不懂生信,做生信的不懂分子,如何让自己走的更远?
工欲善其事,必先利其器,各行各业都有其不二法宝,科研工作亦当如此;
1. 浏览器
最好的浏览器当然是 Google Chrome (不接受任何反驳),简洁无广告净化双眼;再配合其他插件 (http://tiramisutes.github.io/tiramisutes.github.io/2015/07/25/chrome.html) ,只能用如虎添翼来形容;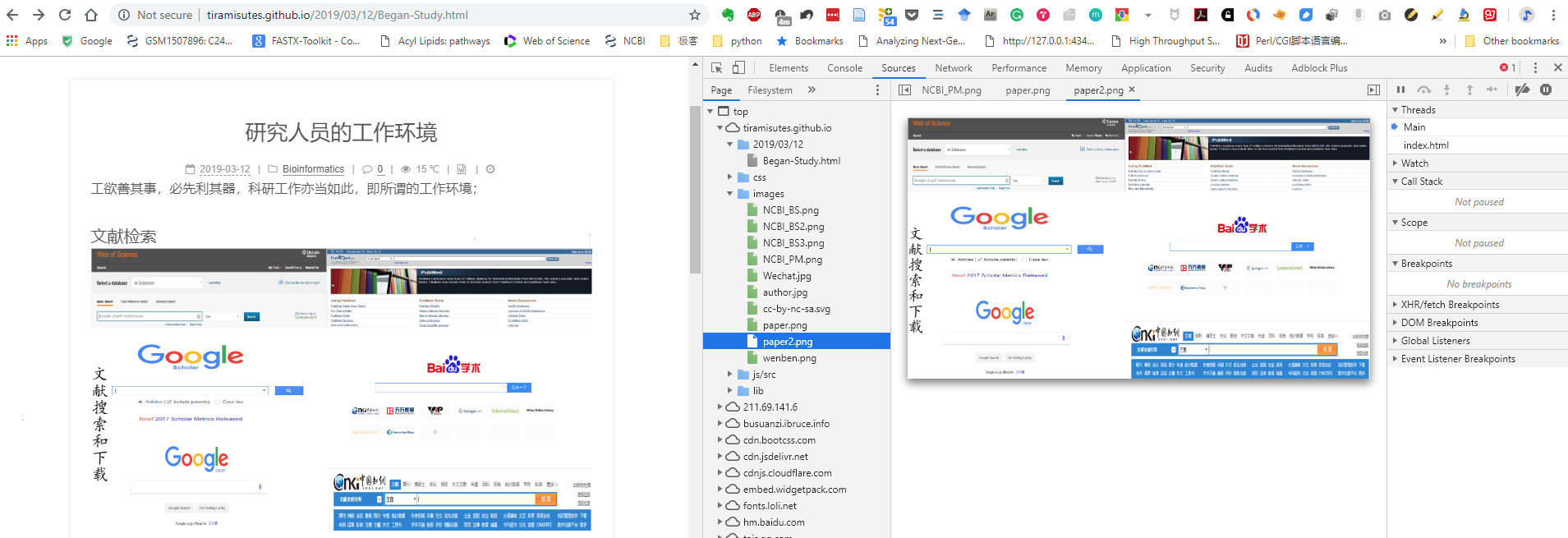
2. 文献检索
最好用的当然也是无所不能的 Google (不接受任何反驳);
实在不行 NCBI 的PubMed也凑合,配合 Scholarscope (http://blog.scholarscope.cn/) 还可以显示期刊领域排名和IF等信息;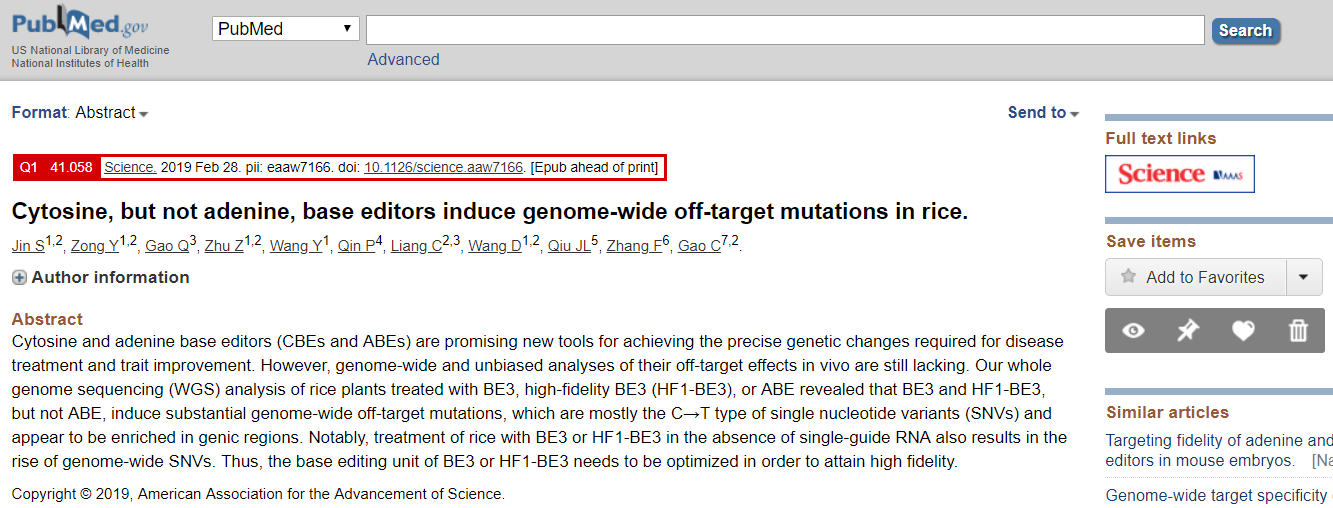
对不起,请再也不要说不会下载文献 (sci-hub.tw);

3. NCBI 数据库
NCBI作为生物类主要数据库之一,查序列再顺手做个blast比对也是常规操作,看完下图让你以后的比对有更多的选择;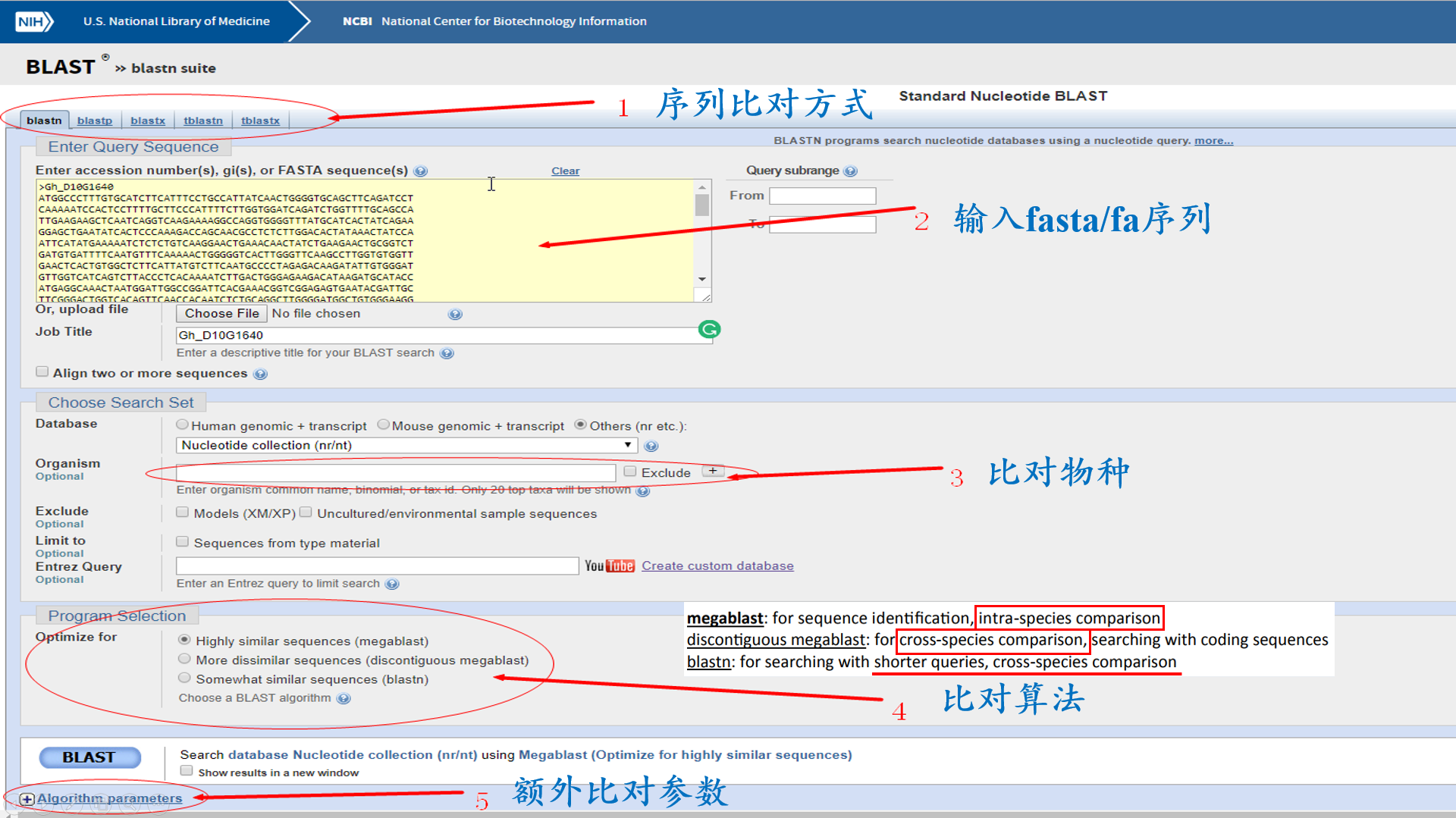
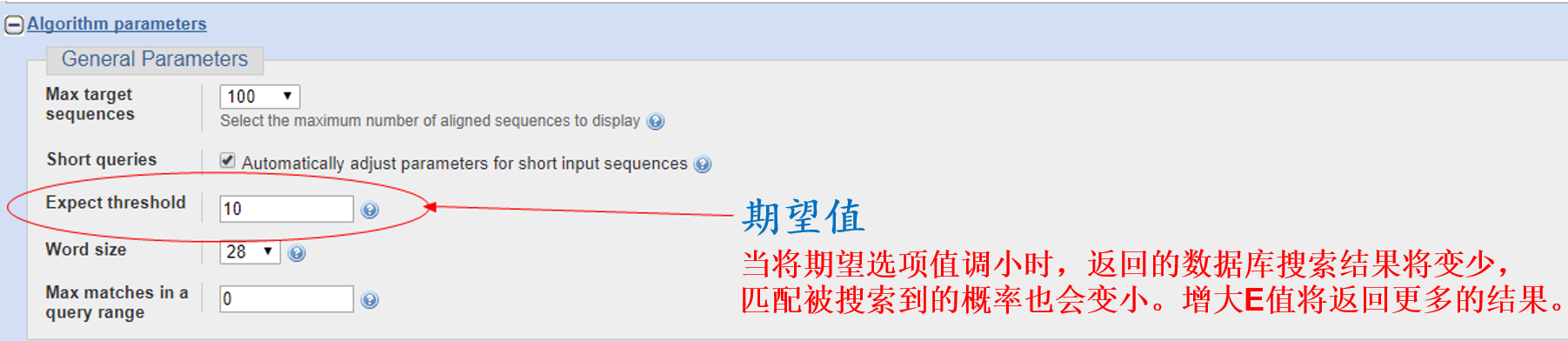
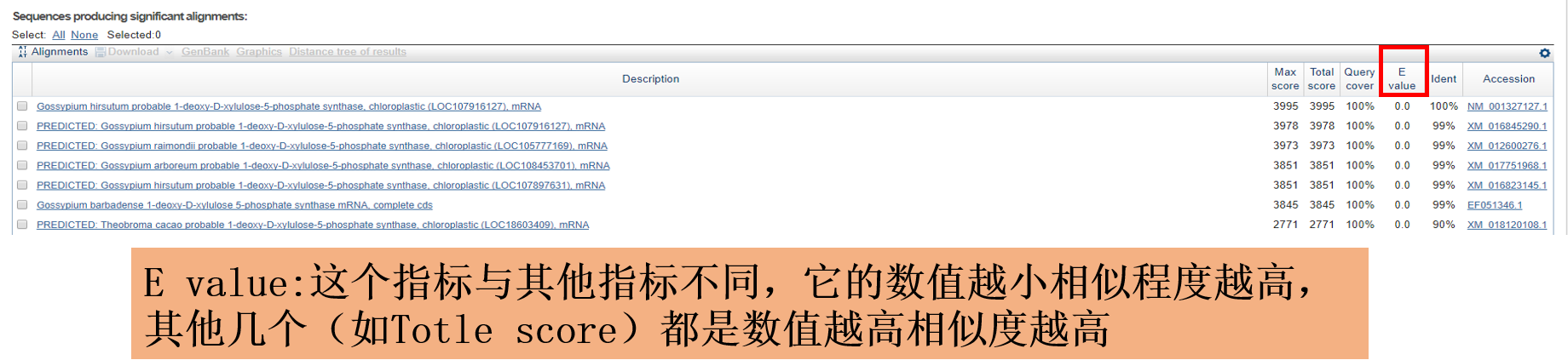
4. 文献管理
文献最主要两种使用方式就是阅读和写文章时的引用,业内巨头EndNote大家很熟悉,但开源的Zotero其实可以弥补非正版EndNote的缺陷;身处云的时代怎么能没有同步功能,完全没有安全感;Zotero网页点击即可保存文献,文献管理、同步、甚至手机端阅读样样精通;
5. 文本编辑
fasta等序列文件的察看,没有比较就没有伤害,同样的文件不同的打开方式,格然不同的阅读效果,轻量可打开绝大多文件类型且支持功能扩展的 Sublime (http://www.sublimetext.com/) 值得你拥有;
6. 数据安全
有备无患,辛辛苦苦得来的实验数据实时备份准没错,硬盘和云端双保险最佳;免费版坚果云和会员版百度云可供选择;当然免费的经特殊处理也可以,详细见:Linux和windows定时备份数据到百度云盘
我是分隔线
好了,如果你看到这里了,可选择性的配备你的工作站,开始接下来的进阶👇
7. 序列比对
windows基础版推荐简单方便且直观的 DNAMAN;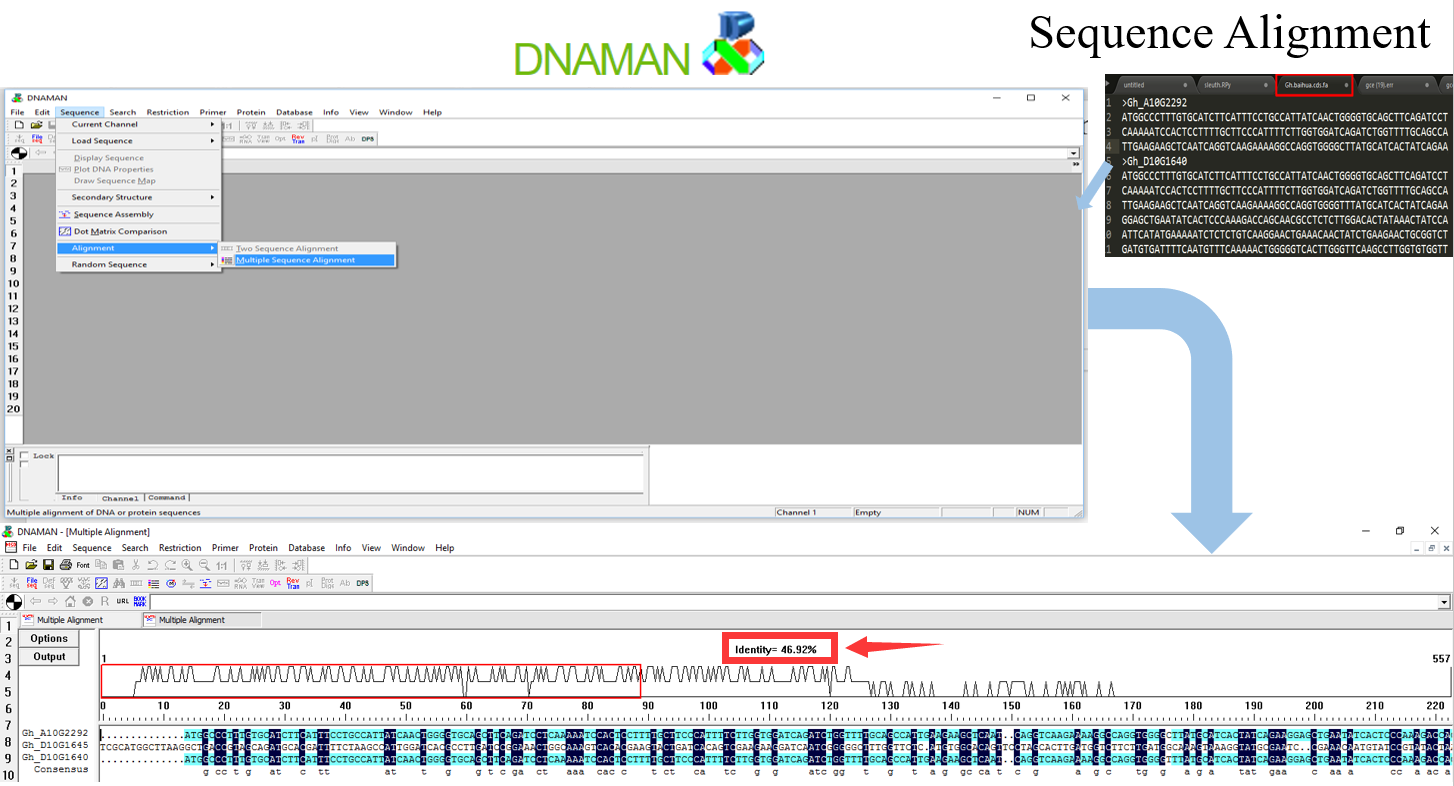
如果你序列太多,则最好Linux下计算,软件首选目前公认比对精确度最高的,需要调节的参数比较少的 MAFFT;
8. 进化构建
NJ法,ML法,BI法是目前主流的建树方法,MP法目前相对用得较少,每种方法都有它一定的优点,同时也存在着缺点。因为进化树的构建是一个统计学问题,我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果采用一个适当的方法,那么所构建的进化树就会接近真实的“进化树”,所以对于相同的数据集,推荐用两种及两种以上的方法建系统发育树进行分析,互相比照。
| 方法 | 适用范围 | 优点 | 缺点 |
|---|---|---|---|
| 邻接法 neighbor-joining (NJ) | 远缘序列且进化距离不大 | 计算速度快,结果 相对准确 | 序列上的所有位点都被同等对待,而所分析序列的进化距离不能太大 |
| 最大似然法 maximum likelihood (ML) | 跨物种比较 ,有模型有模型的情况下ML是与进化事实最吻合的树 | 很好的统计学基础,大样本时似然法可以获得参数统计的最小方差,在进化模型确定的情况下,ML法是与进化事实吻合最好的建树算法 | 计算量大,耗时时间长;依赖于合适的替代模型。 |
| 贝叶斯法 Bayesian | 大而复杂的数据集 | 具有坚实的数学和统计学基础,可以处理复杂和接近实际情况的进化模型 | 对进化模型敏感,涉及较多的统计学假设和参数 |
| 最大简约法 maximum parsimony (MP) | 所要比较的序列的碱基差别小;对于序列上的每一个碱基有近似相等的变异率; 没有过多的颠换/转换的倾向;所检验的序列的碱基数目较多(大于几千个碱基); | 某些特殊的分子数据如插入、缺失等序列 | 只适于序列数目N≤12。存在较多回复突变或平行突变时,结果较差。推测的树不是唯一的,变异大的序列会出现长枝吸引而导致建树错误 |
进化树分类通常为有根树和无根树,有根树反映树上物种(基因)进化的时间顺序,无根树只反映分类单元间的距离,不涉及共同祖先问题;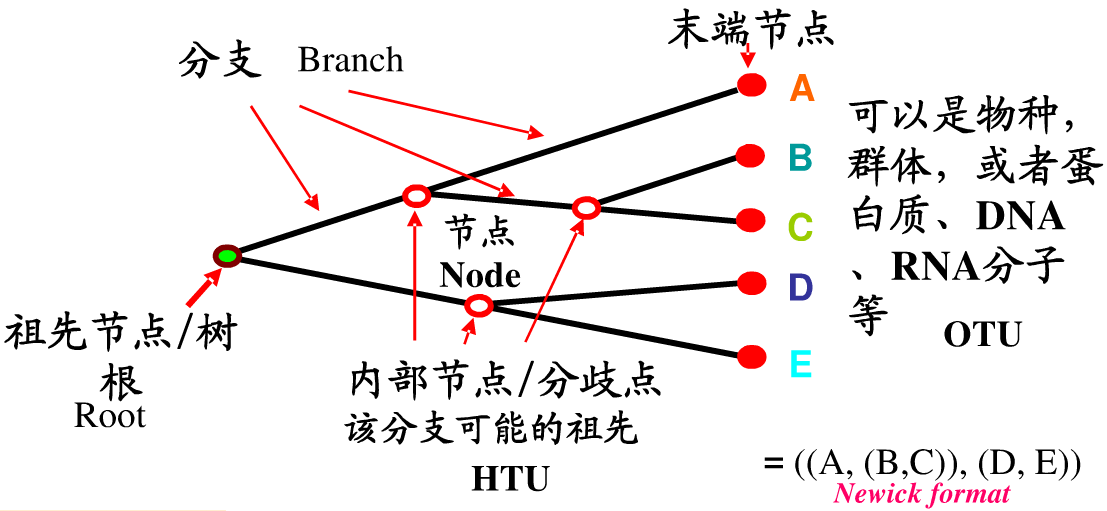
Bootstrape是常用的系统进化树评估优化方法,树枝上的Bootstrape检验值表示该分支通过检验的次数占总次数的百分比,其值大小(>70)反映树枝的可信度;
windows下推荐MEGA,比对进化一步到位,支持最常用的ML和NJ算法;
如果你序列太多,则最好Linux下计算,软件推荐Phylip,RaxML,IQ-tree和MrBayes;
美化推荐iTOL,反正用过的都说好;
