简介(From Wikipedia, the free encyclopedia)
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
ChIP-seq is used primarily to determine how transcription factors and other chromatin-associated proteins influence phenotype-affecting mechanisms. Determining how proteins interact with DNA to regulate gene expression is essential for fully understanding many biological processes and disease states.
流程
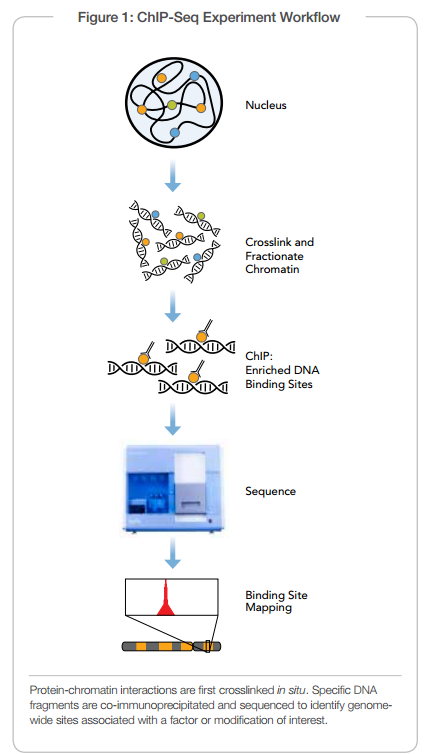
ChIP
The ChIP process enriches specific crosslinked DNA-protein complexes using an antibody against the protein of interest. For a good description of the ChIP wet lab protocol see ChIP-on-chip. Oligonucleotide adaptors are then added to the small stretches of DNA that were bound to the protein of interest to enable massively parallel sequencing.
Sequencing
After size selection, all the resulting ChIP-DNA fragments are sequenced simultaneously using a genome sequencer. A single sequencing run can scan for genome-wide associations with high resolution, as opposed to large sets of tilingarrays required for lower resolution ChIP-chip.
中文解释
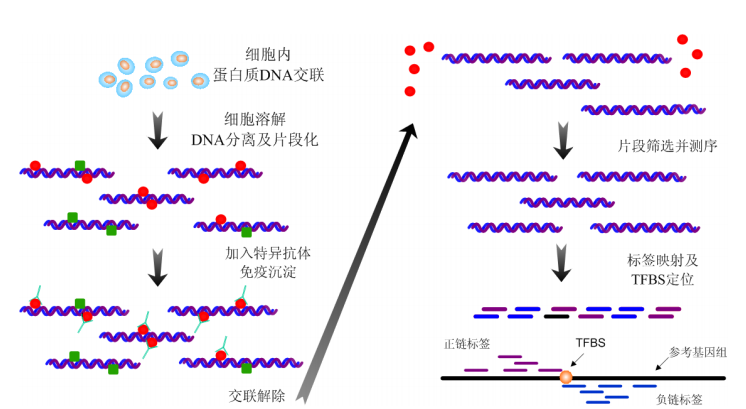
ChIP 测序的基本原分为 ChIP 和测序两个步骤。ChIP 是英文 Chromatin immunoprecipitation 的缩写,即染色质免疫沉淀,其步骤包括:细胞内蛋白质和 DNA 的交联、DNA 分子分离及片段化、免疫沉淀和解除交联。测序就是对解除交联后的 DNA 片段进行测序,制备文库时也包括连接测序接头和片段筛选等步骤。由于免疫沉淀的 DNA 片段在蛋白质结合区域周围富集,因此识别蛋白质结合区域转化为检测测序标签富集的区域。在信息处理中,这是一个信号检测的问题。在转录因子 ChIP 测序数据分析中,由于测序标签还会在转录因子结合位点周围形成分布,通过对分布的统计建模分析,可从数据中精确定位结合位点。
Computational analysis
The read count data generated by ChIP-seq is massive. It motivates the development of computational analysis methods. To predict DNA-binding sites from ChIP-seq read count data, peak calling methods have been developed. The most popular method is MACS which empirically models the shift size of ChIP-Seq tags, and uses it to improve the spatial resolution of predicted binding sites.[10]
Another relevant computational problem is Differential peak calling, which identifies significant differences in two ChIP-seq signals from distinct biological conditions. Differential peak callers segment two ChIP-seq signals and identify differential peaks using Hidden Markov Models. Examples for two-stage differential peak callers are ChIPDiff[11] and ODIN.
具体数据分析详细见:http://www.plob.org/2012/09/29/3760.html
Contribution from :
https://en.wikipedia.org/wiki/ChIP-sequencing
http://www.illumina.com/documents/products/datasheets/datasheet_chip_sequence.pdf
http://bioinfo.au.tsinghua.edu.cn/member/xwang/files/Thesis_XiWang_OL.pdf
