Integrative Genomics Viewer (IGV)作为一个高性能的可视化工具,可以交互式的察看综合的基因组相关数据,也友好的支持多种数据类型,自然是生信工作者必须使用的利器之一。官网也提供了很详细的使用讲解,这里仅是根据我目前需要学习摘录部分做的整理,后面有时间在做其他整理。
1. 输入数据准备
IGV可以导入多种类型的数据,详见下文的数据导入介绍,此处主要说的是排序后的 bwa 的比对文件:bowtie2/BWA + samtools (samtools view>samtools sort>samtools index) 处理结果或RNA-seq的 Tophat结果;
2. 主界面
2.1 基础主界面
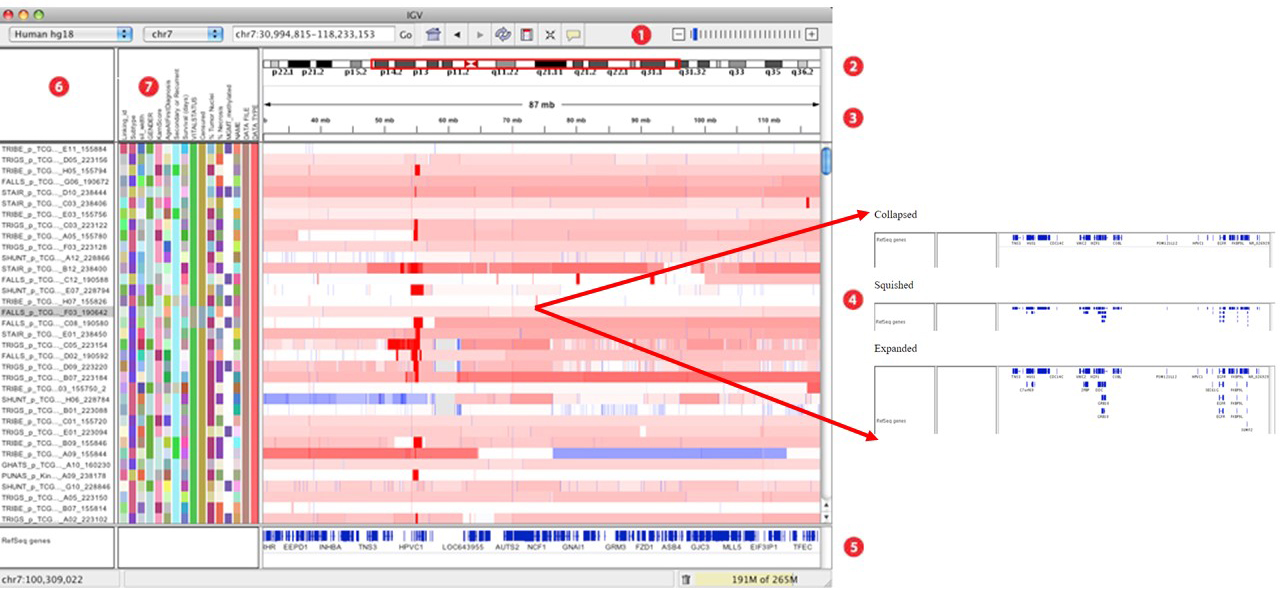
- 工具栏;
- 红框表示显示当前染色体的相应区域;
- 刻度线表示所处位置坐标;
- tracks区域,也即 Alignment Track区;主要的信息区,通常会显示甲基化、基因表达、拷贝数、杂合性缺失(Loss of Heterozygosity)、突变等信息;对应的有三种显示形式:Collapsed、Squished 和 Expanded;
- 特征显示区;蓝色粗线—外显子区域,细线内含子区域,空白—基因间隙;
- 列出 Track names,即导入的比对结果名称;
- 属性面板;
2.2 结果界面说明
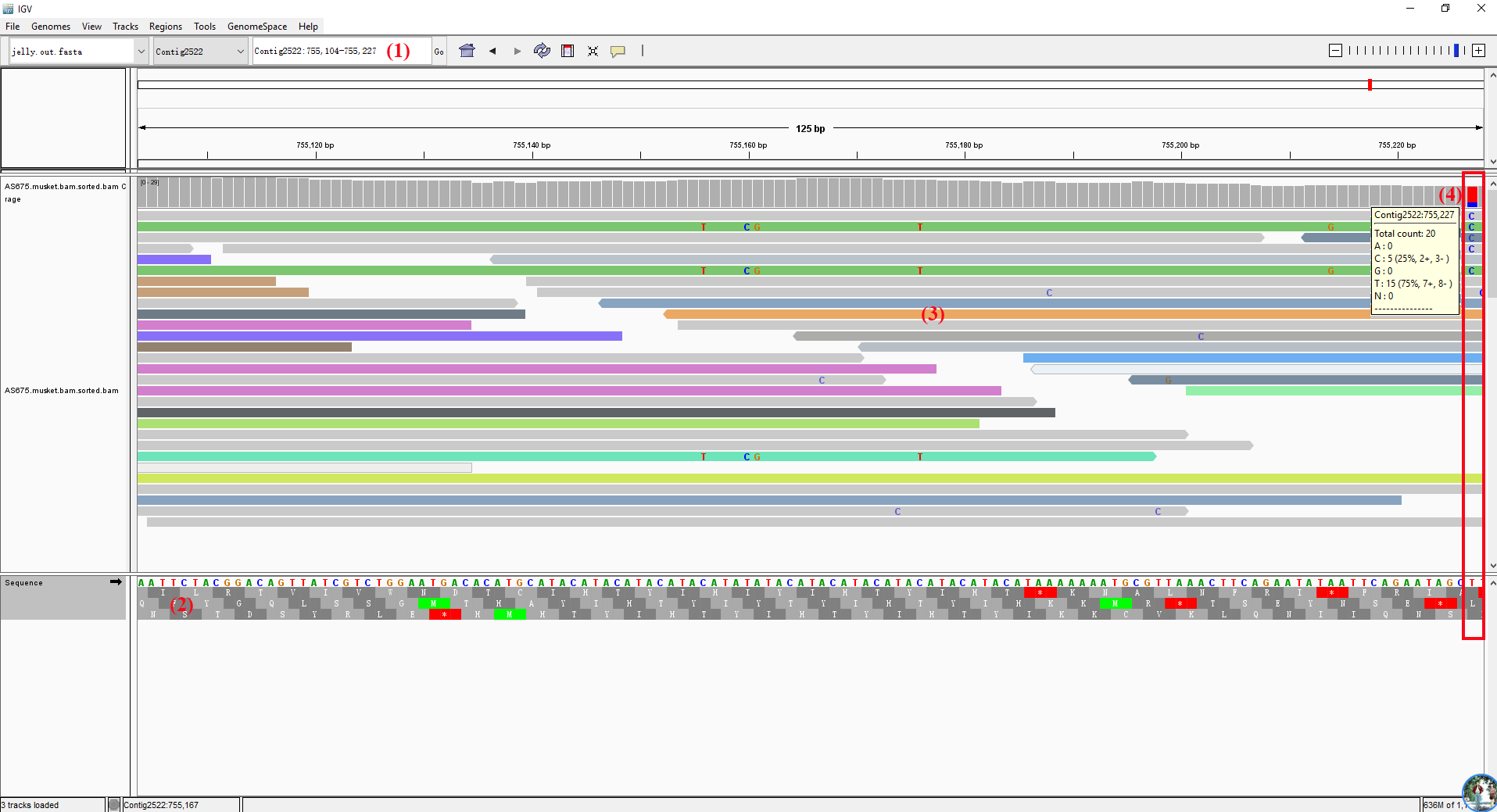
- (1) 处可手动输入想要察看的染色体/contigs/scaffolds编号,然后回车察看;
- (2) 处是参考序列对应的核酸序列,其中四种核酸分别用不同的颜色表示:(A, C, G, T),下面为对应的翻译的氨基酸序列,甲硫氨酸(M)用绿色表示,终止密码子(*)红色星号表示;当右上角的标尺足够大时此区域才会显示;
- (3) 处不同颜色条表示排序方式,鼠标停留在此处右键选择
<Color alignments by>可选取不同的颜色形式;同时每一个长条对应的序列和比对信息可以鼠标右键选择来拷贝;每一个长条都是由一系列的核酸序列组成,可通行<Show all bases>来显示;比对的reads长条也可通过成对的形式显示; - (4) 处鼠标停留时会显示此处碱基统计信息,例如在此处显示为红蓝色,红色是T,蓝色是C,红色方块大于蓝色,表示所有比对到这一位置的序列中这一位点碱基是T的序列大于C的,即C可能是突变;当导入数据为比对的bam数据时,此处所在区域为 Coverage Track,
3. 数据导入
当数据通过 <File> 导入时,IGV通过导入文件的扩展名来确认数据格式 (file format),进而确定数据类型 (data type),再确定数据展现的 Track 形式 (track default display options);如下所示(此默认值均可修改):
4. 察看序列比对结果
- 可通过
View >>Preferences >>Alignments面板设置相关参数; - 在 Track 区不进行
Color alignments by的情况下,alignments 只有亮灰和白色两种长条,其中白色的比对质量为零 (mapping quality equal to zero); - 插入:用紫色的
I或红色的I(当插入的碱基数多余预设的阀值时)表示;鼠标停留察看详细的插入碱基情况; - 缺失:黑条表示;

Sort alignments by可对Track区域进行排序,如想返回最初结果则选择Re-pack alignments即可;- 默认情况下
Track Alignments区以左图紧凑的单个 reads 的形式展示,通过View as pairs可成对显示,且中间以细线连接 (右图);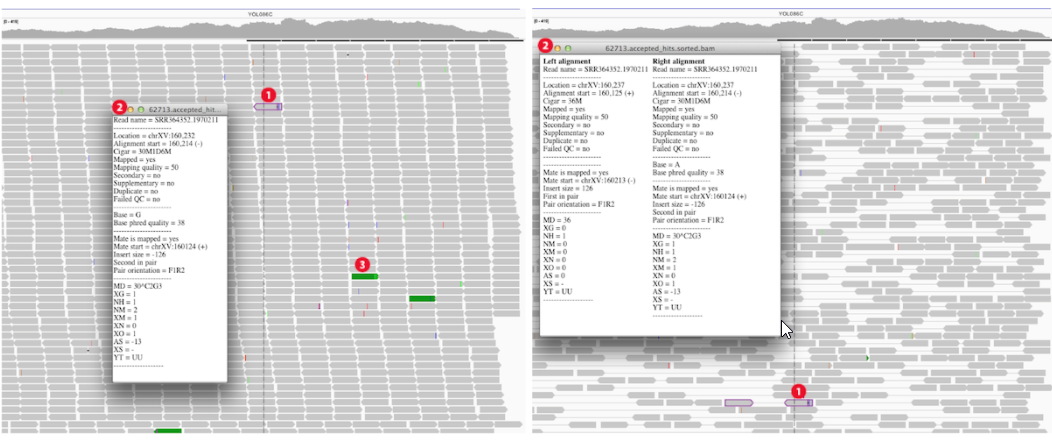
在左图中按住Ctrl键鼠标左击某一个长条 (a read),将以相同的彩色颜色显示出与其配对 (paired mate) 的另一条 read。黑色的表示没有与之配对的另一条read。选中一条 read 后右键Go to Mate将会跳转到与其配对 (paired mate) 的另一条 read。If the paired reads have a large insert size, the paired mate will not be highlighted. 右键选择Clear Selections来清除所有选择的reads。同时注意到不同reads会用不同的颜色表示 (蓝色:插入大小小于期望值;红色:插入大小大于期望值;绿色、青色、深蓝色:倒置、重复、易位事件),更多详情见:Interpreting Color by Insert Size 和 Interpreting Color by Pair Orientation;低分辨率下在 Track Alignments 区域选择Color alignments by >> insert size and pair orientation时比对的reads会显示不同的颜色 (Red have larger than expected inferred sizes, and therefore indicate possible deletions; Blue have smaller than expected inferred sizes, and therefore indicate insertions;实心灰代表比对质量比较高的测序片段,空心灰代表比对到此处的测序片段也可以比对到其他位点。),高分辨率下,可以精确到每个位点的碱基类型:当比对序列上与参考基因组相同的超过80%时,用灰色表示;否则用红色-T,蓝色-C,绿色-A,橙色-G;Translocations on the same chromosome can be detected by color-coding for pair orientation, whereas translocations between two chromosomes can be detected by coloring by insert size. Paired-end alignment tracks时 (View as pairs),右键选择View mate region in split screen可分隔显示;可实现多个分隔;在下图①处右键选择Switch to standard view或鼠标左键双击可返回单个分区;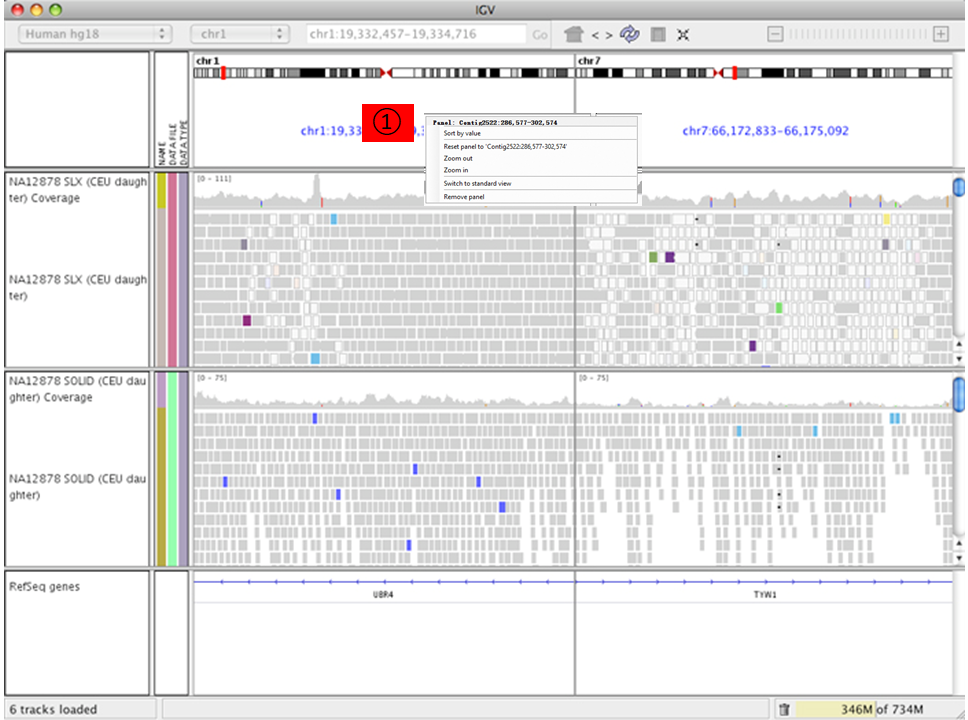
5. 察看可变剪切情况
Loaded junctions data in the standard
.bedformat (例如TopHat’s “junctions.bed”等输出文件);1
2
3
4
5
6
7|-- accepted_hits.bam
|-- accepted_hits.bam.bai
|-- deletions.bed
|-- insertions.bed
|-- junctions.bed
|-- unmapped.bam
`-- unmapped.bam.bai载入RNA-seq比对基因组bam文件
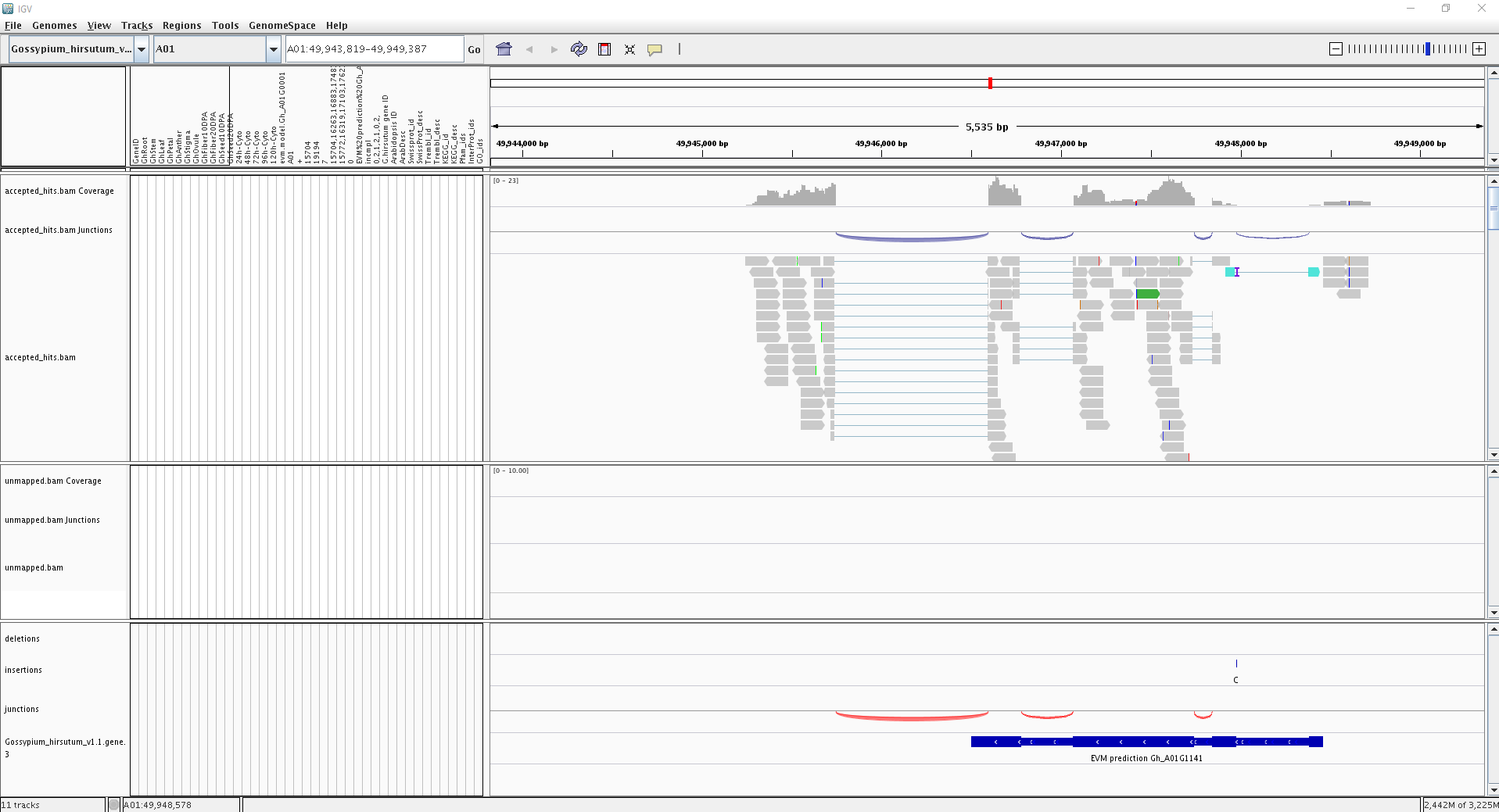
图示说明:
- 红色向上弧形表示可变剪切基因位于正链,蓝色向下为负链;
- 弧形的高度和厚度(thickness)与reads覆盖度成比例;
6. 察看变异
6.1 Mutation Files:MAF (mutation annotation format) and MUT (mutation)文件;
6.2 VCF Files

 |
Each bar across the top of the plot shows the allele fraction for a single locus. |
|---|---|
 |
The genotypes for each locus in each sample. Dark blue = heterozygous, Cyan = homozygous variant, Grey = reference. Filtered entries are transparent. |
7. 参考资料
IGV应用教程
