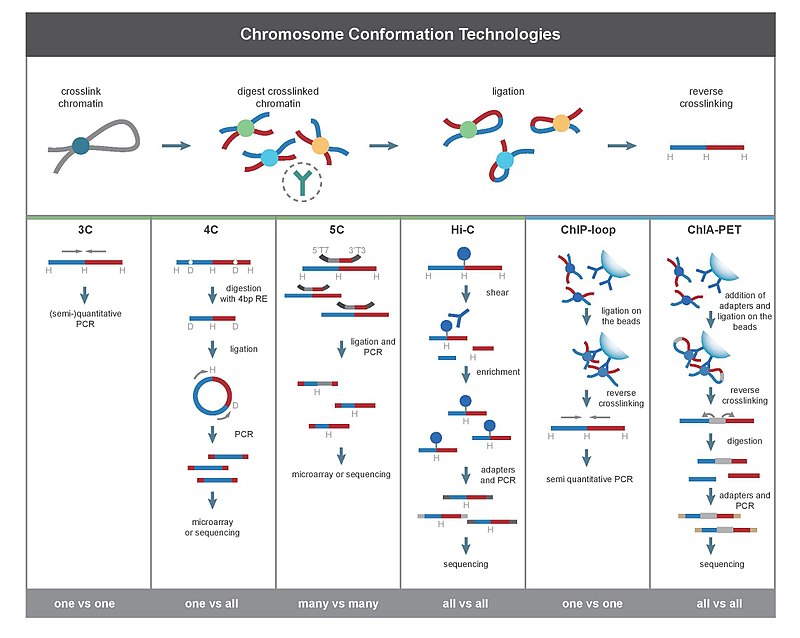
零、常识介绍
Mammalian genomes are spatially organized into compartments, topologically associating domains (TADs), and loops to facilitate gene regulation and other chromosomal functions.
3D interactions mostly occur within chromosomes (cis) rather than between chromosomes (trans), all methods detected more cis than trans interactions.
一、鉴定染色体交互 (identify chromatin interactions)
Chromatin interactions are contacts between regions far from each other on the linear DNA sequence but close in 3D space;
| Function | Method | vailability | Programming language |
|---|---|---|---|
| Chromatin interactions | Fit-Hi-C | http://noble.gs.washington.edu/proj/fit-hi-c | Python |
| Chromatin interactions | GOTHiC | http://bioconductor.org/packages/release/bioc/html/GOTHiC.html | R |
| Chromatin interactions | HOMER | http://homer.ucsd.edu/homer/interactions/HiCmatrices.html | Perl, R |
| Chromatin interactions | HIPPIE | http://wanglab.pcbi.upenn.edu/hippie/ | Python, Perl, R |
| Chromatin interactions | diffHic | https://bioconductor.org/packages/release/bioc/html/diffHic.html | R, Python |
| Chromatin interactions | HiCCUPS | https://github.com/theaidenlab/juicer/wiki/Download | Java |
| TADs | HiCseg | https://cran.r-project.org/web/packages/HiCseg/index.html | R |
| TADs | TADbit | https://github.com/3DGenomes/TADbit | Python |
| TADs | DomainCaller | http://chromosome.sdsc.edu/mouse/hi-c/download.html | Matlab, Perl |
| TADs | InsulationScore | https://github.com/dekkerlab/crane-nature-2015 | Perl |
| TADs | Arrowhead | https://github.com/theaidenlab/juicer/wiki/Download | Java |
| TADs | TADtree | http://compbio.cs.brown.edu/projects/tadtree/ | Python |
| TADs | Armatus | https://github.com/kingsfordgroup/armatus | C++ |
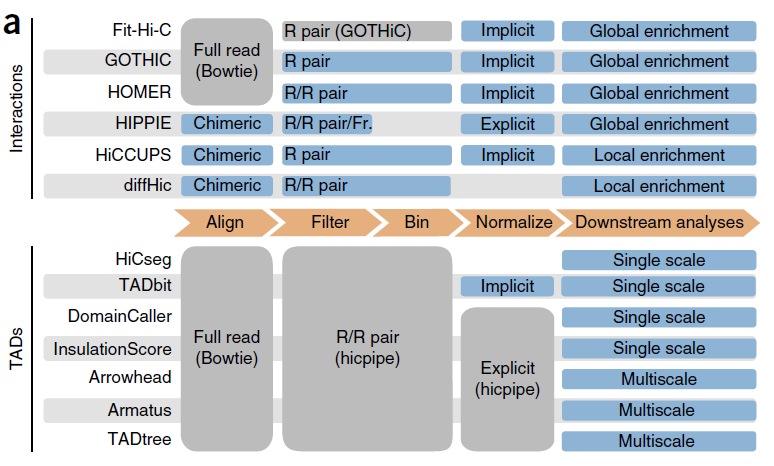
【上述图表来源于:Comparison of computational methods for Hi-C data analysis】
The total number of interactions called by each method increased with the number of reads retained by the filtering step for all tools at any resolution, although the rate of increase varied from tool to tool.
二、Topologically Associating Domains (TADs)
TADs are structural domains consisting of chromatin regions that are highly self-interacting but have limited interaction with regions in other domains;
TADtree: an algorithm the identification of hierarchical topological domains in Hi-C data
Arrowhead: for finding contact domains
Used bin size (resolution) of at least 40 kb for TAD calling;
1 | cd /public/home/cotton/software/juicer/data |
三、Compartments
- CscoreTool: fast Hi-C compartment analysis at high resolution
- Eigenvector: used to delineate compartments in Hi-C data at coarse resolution
The genome-wide chromosome conformation capture (Hi-C) has revealed that the eukaryotic genome can be partitioned into A and B compartments that have distinctive chromatin and transcription features.
The current method for calculating A/B compartments is based on the Principal Component Analysis (PCA) of the normalized Hi-C interaction matrix (Lieberman-Aiden et al., 2009). The first eigenvector (Principal Component 1, PC1) of the correlation matrix is then defined as the compartment score, and genomic windows with positive or negative compartment scores are defined as A or B compartment, respectively.
1 | cd //home/cotton/software/juicer/data |
hic 文件来源于 3D DNA 流程或 java -Xmx2g -jar juicebox_tools.jar pre;
四、Chromatin loops
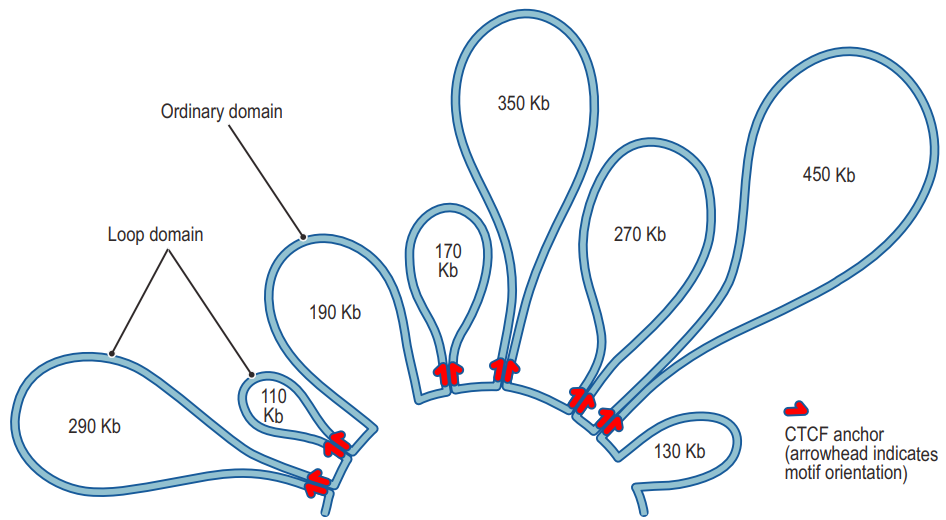
- Chromatin loops in gene regulation
- CTCF-Mediated Chromatin Loops between Promoter and Gene Body Regulate Alternative Splicing across Individuals30488-X)
- Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins
HiCCUPS is an algorithm for finding chromatin loops.
HiCCUPS 算法包含在 juicer 软件中,可按照如下手动单独运行(有GPU节点使用):1
2
3
4java -Xmx2g -jar ./CPU/juicer_tools.jar hiccups -h
java -Xmx2g -jar ./CPU/juicer_tools.jar hiccups /public/home/cotton/software/3d-dna/xzp/Hs1.split.hic all_hiccups_loops
java -Xmx2g -jar ./CPU/juicer_tools.jar hiccups -r 40000 -p 1 -i 3 -f 0.1 -d 80000 --ignore_sparsity /public/home/cotton/software/3d-dna/xzp/Hs1.split.hic hiccups_40kb
五、软件性能比较
过滤步骤
- HiCCUPS retained the largest number of aligned reads, although it is worth noting that HiCCUPS filters only PCR duplicates without discarding other potential artifact reads.
- diffHic filtered the highest proportion of aligned reads in most data sets (from 27% to 94%, depending on the data set); but, given its higher alignment rate, still retained a large number of reads.
Identification of chromatin interactions
- GOTHiC called the highest number of cis interactions;
- diffHic found the largest number of trans interactions;
- HiCCUPS, which aggregates nearby peaks into a single interaction, identified fewer interactions than all other tools.
- For interaction callers, HOMER and HiCCUPS yielded the highest proportion of interactions with a potential biological significance—although the potential of HiCCUPS could be fully exploited only in the analysis of very high-resolution data sets.
Distance between the interacting points in cis
- GOTHiC found interactions at shorter mean distance at both 5- and 40-kb resolutions;
- At 5 kb, Fit-Hi-C called interactions at an average distance of more than 10 Mb; which was expected, as Fit-Hi-C is designed to call midrange interactions.
- At low resolution, GOTHiC had the highest concordance, most likely because it called a large number of short-range interactions in every sample replicate.
- At high resolution, the interactions found by HiCCUPS were the most conserved among replicates.
- At 5kb resolution, HiCCUPS and HOMER called the highest proportion of promoter–enhancer interactions, although not the highest absolute number.
cis interaction 正确性和敏感性
- GOTHiC recovered the largest number of true-positive interactions. HOMER and Fit-Hi-C performed comparably to GOTHiC, although they called a smaller number of total interactions.
- In high-resolution data sets, diffHic recalled the highest number of true positives, although HOMER identified more true positives than any other tool at comparable numbers of called interactions.
- The highest sensitivity was achieved by Fit-Hi-C.
Identification of topologically associating domains
- The number of TADs did not increase with the number of reads retained after filtering for all tools, with the exception of Arrowhead.
- At 40-kb resolution, TADtree called the largest (7,638) and Arrowhead the smallest (636) number of TADs. Conversely, at 1-Mb resolution, InsulationScore returned the largest number of TADs.
- Note that some methods (HiCseg, TADbit, InsulationScore) partition chromosomes in a continuous set of TADs, whereas the others allow gaps between TADs. Arrowhead and TADtree, which adopt multiscale approaches, returned nested TADs.
- TADs identified by HiCseg were also the most reproducible when using the overlap coefficient.
六、分析流程

