用SOAPdenovo对Illumina paired-end进行基因组组装时需要配置文件,其中要填写每个文库的average insert size,那么如何进行average insert size大小的评估呢?
文库类型
对于基因组文库我们一般会建小库(<1K)的paired-end reads和大库的mate-pair reads,二者最主要的区别就是reads1和reads2的方向和之间的间隔大小。
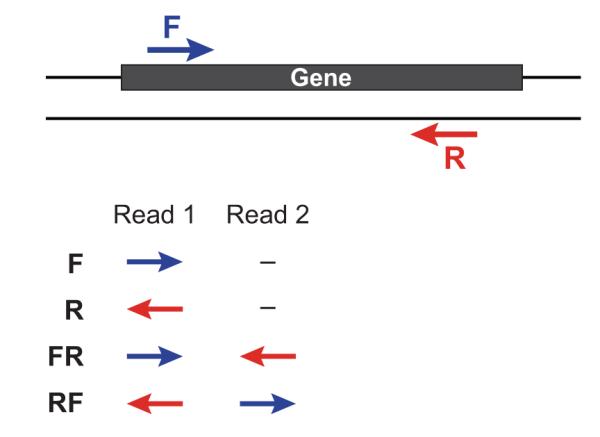
现在绝大部分的主流软件都是支持将paired-end reads进行比对的,那么 mate-pair reads如何处理呢,即 mate-pair reads如何做比对?请参考我的另一篇博文 Mate-pair Reads Alignment
Insert Size
首先,什么是Insert Size呢?
对于paired-end reads来说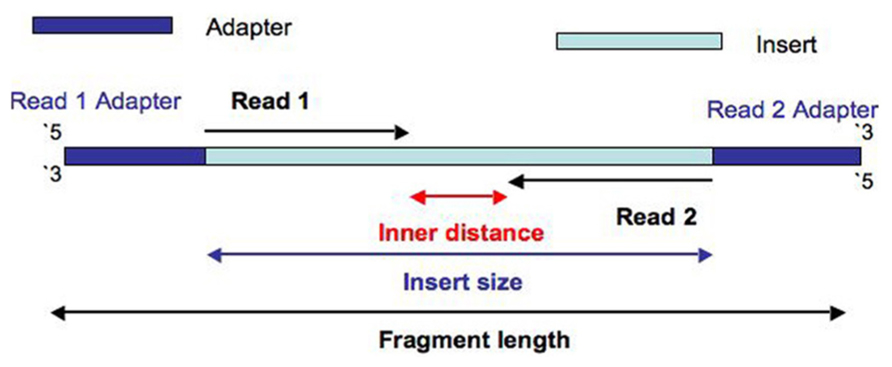
对于mate-pair reads来说其reads1和reads2方向指向外面,其插入大小统计需要格外注意。
基于bwa比对log文件统计插入大小
通过观察bwa软件的输出log文件发现其对每一个pair-end reads分4次读入([M::main_mem] read 2613712 sequences (200000105 bp)…),对于每一次的读入会对reads进行统计如下:
由红框标出发现占主要比例的是RF reads,进一步往下寻找analyzing insert size distribution for orientation RF…就可得到其平均插入大小。
基于比对的sam文件统计插入大小
R计算
1 | $ cat sample.sam | cut -f9 > initial.insertsizes.txt |
可见R计算过程选择过滤掉小于等于15%和大于等于85%的值来计算平均插入大小;
awk计算
1 | awk '{ if ($9 > 0) { N+=1; S+=$9; S2+=$9*$9 }} END { M=S/N; print "n="N", mean="M", stdev="sqrt ((S2-M*M*N)/(N-1))}' sample.sam |
awk选择全部大于0的值计算平均插入大小;
基于sorted.bam文件
qualimap
1 | qualimap bamqc -bam sample.sorted.bam --java-mem-size=300G -c -nw 400 -hm 3 -outdir /resulted |
CollectInsertSizeMetrics
1 | java -jar CollectInsertSizeMetrics.jar \ |
问题
现对同一个比对产生的sam/bam文件用上述4种方法计算得出结果如下:
R :mean insert size = 260.577232343453”,”standard deviation = 27.4153198790634”;
awk: mean=250.826, stdev=51.7005;
qualimap:mean insert size = 250.8258,std insert size = 51.7005;
CollectInsertSizeMetrics:MEAN_INSERT_SIZE=260.963523,STANDARD_DEVIATION=42.809159。
qualimap 和 CollectInsertSizeMetrics 都是java封装的软件,看不到其具体计算方法,根据以上计算结果可以看出CollectInsertSizeMetrics的计算原理应该和R的一样需要过滤掉数据,qualimap和awk中发一样,所以问题最后就归结为是否需要首先过滤数值再计算平均插入大小?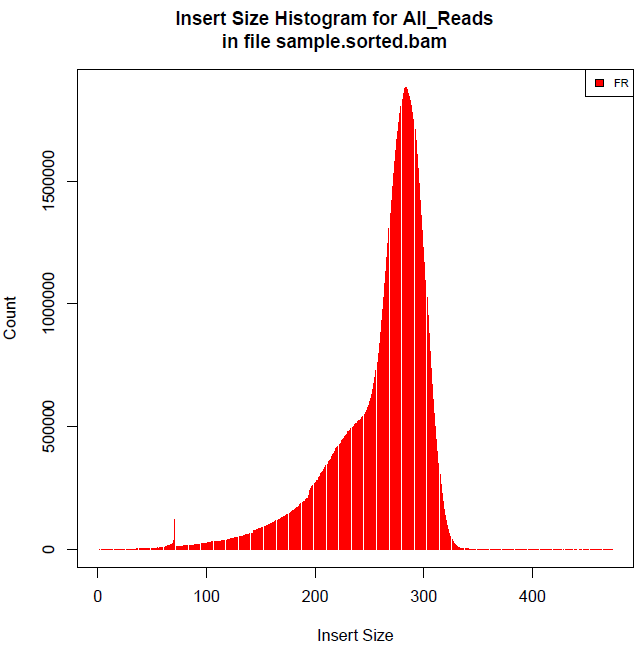
在R中计算时对数据a.v做正态性检验lillie.test(a.v)1
2
3
4
5
6
7> library("nortest")
> lillie.test(a.v)
Lilliefors (Kolmogorov-Smirnov) normality test
data: a.v
D = 0.1541, p-value < 2.2e-16
可以看出其插入大小分布不是呈正态分布,综合考虑后还是按照R的计算结果为准。
参考资料
Question: Estimate Insert Size In Paired-End/Mate-Pair
Question: What is the difference between a Read and a Fragment in RNA-seq?
Paired-end read confusion - library, fragment or insert size?
