文库类型
对于基因组文库我们一般会建小库(<1k)的**paired-end reads="" (l-=""> <-R) 和大库的 mate-pair reads(<-L R->),二者最主要的区别就是reads1和reads2的方向和之间的间隔大小。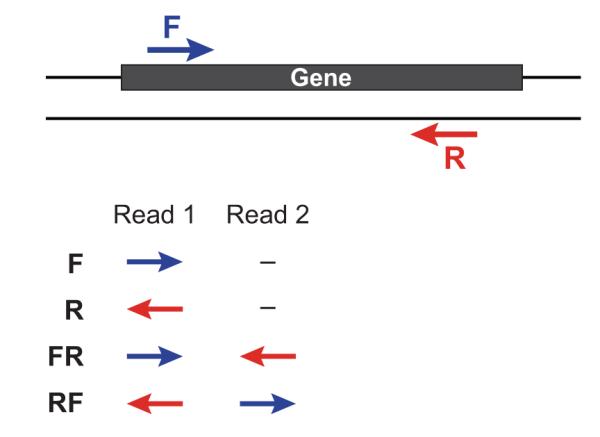
现在绝大部分的主流软件都是支持将paired-end reads进行比对的,那么 mate-pair reads如何处理呢,即 mate-pair reads**如何做比对?
reverse complement
When done standard Illumina MP preps, reverse complemented with fastx-toolkit and aligned with standard parameters using bwa/bowtie.
fastx-toolkit reverse complement
FASTQ/A Reverse Complement
$ fastx_reverse_complement -h usage: fastx_reverse_complement [-h] [-r] [-z] [-v] [-i INFILE] [-o OUTFILE] version 0.0.6 [-h] = This helpful help screen. [-z] = Compress output with GZIP. [-i INFILE] = FASTA/Q input file. default is STDIN. [-o OUTFILE] = FASTA/Q output file. default is STDOUT.
bowtie2
也可通过设置bowtie2的—fr/—rf/—ff、-I、-X参数来进行比对。
Aligning pairs
A “paired-end” or “mate-pair” read consists of pair of mates, called mate 1 and mate 2. Pairs come with a prior expectation about (a) the relative orientation of the mates, and (b) the distance separating them on the original DNA molecule. Exactly what expectations hold for a given dataset depends on the lab procedures used to generate the data. For example, a common lab procedure for producing pairs is Illumina’s Paired-end Sequencing Assay, which yields pairs with a relative orientation of FR (“forward, reverse”) meaning that if mate 1 came from the Watson strand, mate 2 very likely came from the Crick strand and vice versa. Also, this protocol yields pairs where the expected genomic distance from end to end is about 200-500 base pairs.
Paired-end options
-I/—minins
The minimum fragment length for valid paired-end alignments. E.g. if -I 60 is specified and a paired-end alignment consists of two 20-bp alignments in the appropriate orientation with a 20-bp gap between them, that alignment is considered valid (as long as -X is also satisfied). A 19-bp gap would not be valid in that case. If trimming options -3 or -5 are also used, the -I constraint is applied with respect to the untrimmed mates.
The larger the difference between -I and -X, the slower Bowtie 2 will run. This is because larger differences bewteen -I and -X require that Bowtie 2 scan a larger window to determine if a concordant alignment exists. For typical fragment length ranges (200 to 400 nucleotides), Bowtie 2 is very efficient.
Default: 0 (essentially imposing no minimum)-X/—maxins
The maximum fragment length for valid paired-end alignments. E.g. if -X 100 is specified and a paired-end alignment consists of two 20-bp alignments in the proper orientation with a 60-bp gap between them, that alignment is considered valid (as long as -I is also satisfied). A 61-bp gap would not be valid in that case. If trimming options -3 or -5 are also used, the -X constraint is applied with respect to the untrimmed mates, not the trimmed mates.
The larger the difference between -I and -X, the slower Bowtie 2 will run. This is because larger differences bewteen -I and -X require that Bowtie 2 scan a larger window to determine if a concordant alignment exists. For typical fragment length ranges (200 to 400 nucleotides), Bowtie 2 is very efficient.
Default: 500.—fr/—rf/—ff
The upstream/downstream mate orientations for a valid paired-end alignment against the forward reference strand. E.g., if —fr is specified and there is a candidate paired-end alignment where mate 1 appears upstream of the reverse complement of mate 2 and the fragment length constraints (-I and -X) are met, that alignment is valid. Also, if mate 2 appears upstream of the reverse complement of mate 1 and all other constraints are met, that too is valid. —rf likewise requires that an upstream mate1 be reverse-complemented and a downstream mate2 be forward-oriented. —ff requires both an upstream mate 1 and a downstream mate 2 to be forward-oriented. Default: —fr (appropriate for Illumina’s Paired-end Sequencing Assay).
