问题描述
Genomic Feature通常包括exon、intron、intergenic region、UpstreamToGene、UTRs等,对于有完整参考基因组物种其一般都有注释文件gff3,但其一般只有mRNA,gene和exon的坐标信息,而我们通常也需要更多的Genomic Feature信息。
解决方案
工具
bedtools
bedtools具体使用讲解见我的另一篇博文:bedtools 使用小结。
特征种类
在gff3文件第三列标注有相应的特征类型,我们可以参看每种特征类型的数量:1
cat XX.gff3 | grep -v "^#" | cut -f3 | sort | uniq -c | sort -k1rn
Remove/merge overlapping exons
在gff3文件中我们发现存在有以下情况:1
2
3
4$ grep -B 5 "89201851" Gossypium_hirsutum_v1.1.gene.gff3 | grep "exon"
A01 EVM exon 89201570 89201851 . - . ID=evm.model.Gh_A01G1441.exon1;Parent=evm.model.Gh_A01G1441
A01 EVM exon 89201852 89201963 . - . ID=evm.model.Gh_A01G1442.exon3;Parent=evm.model.Gh_A01G1442
A01 EVM exon 89202063 89202216 . - . ID=evm.model.Gh_A01G1442.exon2;Parent=evm.model.Gh_A01G1442
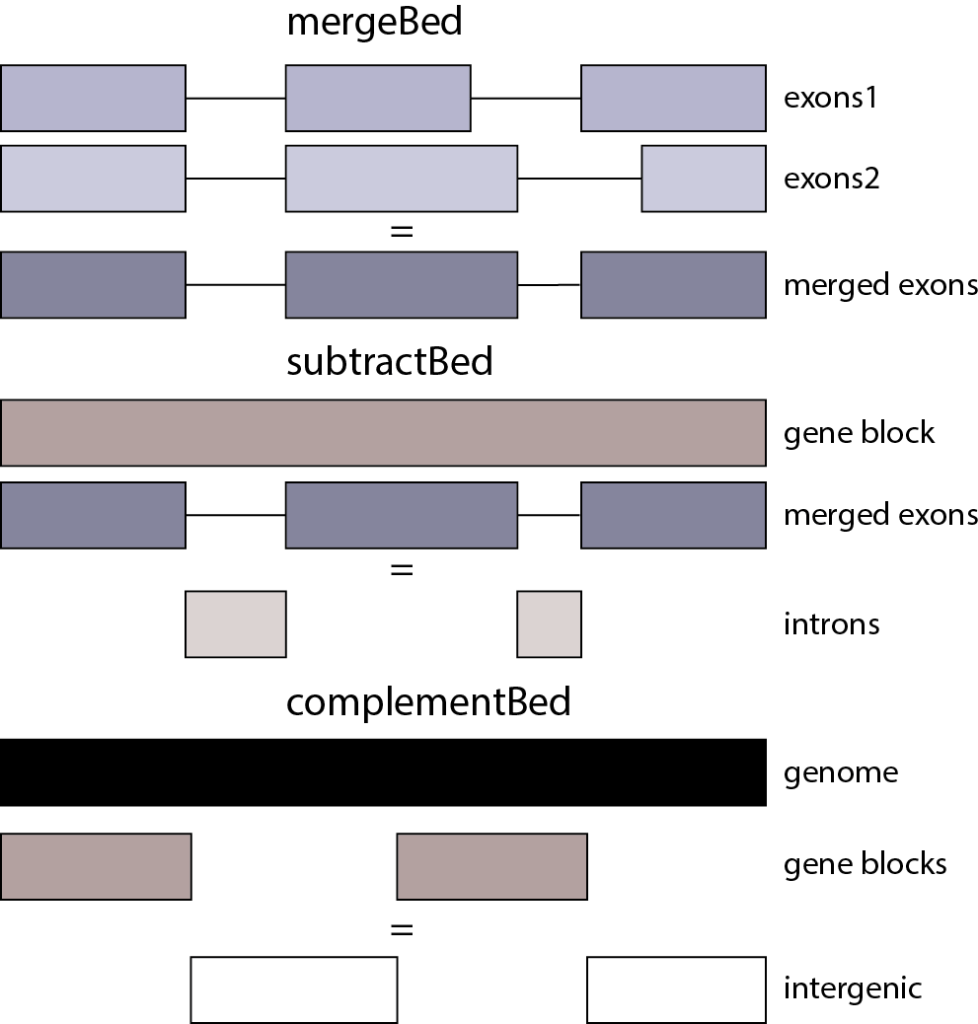
仔细察看发现两个基因Gh_A01G1441和Gh_A01G1442的外显子尽然想连续(第一行终止位置89201851和第二行起始位置89201852),也就是两个连续的基因,在这种情况下有时我们在计算exon时想要将其合并为同一个exon。mergeBed ( Merges overlapping BED/GFF/VCF entries into a single interval):bedtools merge [OPTIONS] -i 1
2
3
4
5
6
7
8
9
10
11
12cat Gossypium_hirsutum_v1.1.gene.gff3 | \
awk 'BEGIN{OFS="\t";} $3=="exon" {print $1,$4-1,$5}' | \
sortBed | mergeBed -i - >merged-exon.gff3
#比较merge前后效果
cat Gossypium_hirsutum_v1.1.gene.gff3 | \
awk 'BEGIN{OFS="\t";} $3=="exon" {print $1,$4-1,$5}' | \
sortBed | diff - merged-exon.gff3
7832,7833c7832
< A01 89201569 89201851
< A01 89201851 89201963
---
> A01 89201569 89201963
Get intron regions
gff3中的intron区就是一个mRNA/gene的exon以外的区域,可通过subtractBed (Removes the portion(s) of an interval that is overlapped by another feature(s)):bedtools subtract [OPTIONS] -a 1
2
3
4
5
6
7
8
9
10
11
12
13
14cat Gossypium_hirsutum_v1.1.gene.gff3 | \
awk 'BEGIN{OFS="\t";} $3=="gene" {print $1,$4-1,$5}' | \
sortBed |subtractBed -a stdin -b merged-exon.gff3 >merged-intron.gff3
#比较 merged-exon.gff3 和 merged-intron.gff3
head -n 4 merged-exon.gff3 merged-intron.gff3
A01 15704 15772
A01 16263 16319
A01 16883 17103
A01 17483 17623
A01 15772 16263
A01 16319 16883
A01 17103 17483
A01 17623 18384
另外一个软件同样可以得到intron:GenomeTools: a comprehensive software library for efficient processing of structured genome annotations,gt gff3 -addintrons Gossypium_hirsutum_v1.1.gene.gff3 >Gh-intron.gff3。
Get intergenic regions

基因间区,即没有基因覆盖的染色体区域。complementBed:bedtools complement [OPTIONS] -i 1
2
3
4
5
6
7
8
9cat Gossypium_hirsutum_v1.1.gene.gff3 | \
awk 'BEGIN{OFS="\t";} $3=="gene" {print $1,$4-1,$5}' | \
sortBed | complementBed -i stdin -g genome.fa.length> merged-intergenic.gff3
#查看 intergenic regions
more Gh-intergenic.gff3
A01 0 15704
A01 19194 22807
A01 24529 36427
A01 36860 40961
上述mergeBed、subtractBed、complementBed操作图解如下: