无参De Nove组装通常用到Trinity软件,组装过程中最重要的两个参数就是--min_kmer_cov 和 --min_glue为组装出高质量结果我们通常需要去尝试用不同的参数,github上也有软件开发者讨论关于这两个参数Optimizing parameters可供参考,其实问题最终也就归结为你是否关心你数据中的低丰度转录本?
此外作者也提供了一系列方法来评估组装质量Transcriptome Assembly Quality Assessment总共列出6种方法可对不同参数的组装结果进行评估,看完后综合总结出其中4种评估方法。
Assessing the Read Content of the Transcriptome Assembly
1 | bowtie2-build ../trinity_out_dir${i}/Trinity.fasta ../trinity_out_dir${i}/Trinity.fasta |
第二步的bowtie2比对序列到组装转录本结果时可选部分数据来比对,这样可大大降低比对耗时。
Full-length transcript analysis for model and non-model organisms using BLAST+
1 | blastall -p blastx -i ./trinity_out_dir${i}/Trinity.fasta -d ${uniprot} -v 1 -b 1 -m 8 -e 1e-5 -a ${cpu} -F F -o uniprot_sprot.fasta_blastx.outfmt8 |
Compute DETONATE scores
RSEM-EVAL软件对于双端reads数据需要提供一个average fragment length值,可参考我的另一篇博文评估文库 Average Insert Size来计算得到此值。1
2
3
4
5
6rsem-eval-estimate-transcript-length-distribution ./trinity_out_dir${i}/Trinity.fasta ./RSEM-EVAL${i}/length_distribution_parameter.txt
rsem-eval/rsem-eval-calculate-score -p 1 \
--transcript-length-parameters ./RSEM-EVAL${i}/length_distribution_parameter.txt \
--paired-end --phred33 --strand-specific ../1.clean.fq ../2.clean.fq\
./trinity_out_dir${i}/Trinity.fasta \
hope-trinity_out_dir${i} 300
评估结果解释见:RSEM-EVAL: A novel reference-free transcriptome assembly evaluation measure。
sample_name.score: stores the evaluation score for the evaluated assembly. The first lines Score the RSEM-EVAL score.BUSCO explore completeness according to conserved ortholog
1 | git clone https://gitlab.com/ezlab/busco.git |
点击BUSCO官网相应图标下载所需数据库。
1
python BUSCO.py -i SEQUENCE_FILE -o OUTPUT_NAME -l LINEAGE -m tran
SEQUENCE_FILE:transcript set (DNA nucleotide sequences) file in FASTA format
OUTPUT_NAME:name to use for the run and temporary files (appended)
LINEAGE:location of the BUSCO lineage data to use (e.g. fungi_odb9)
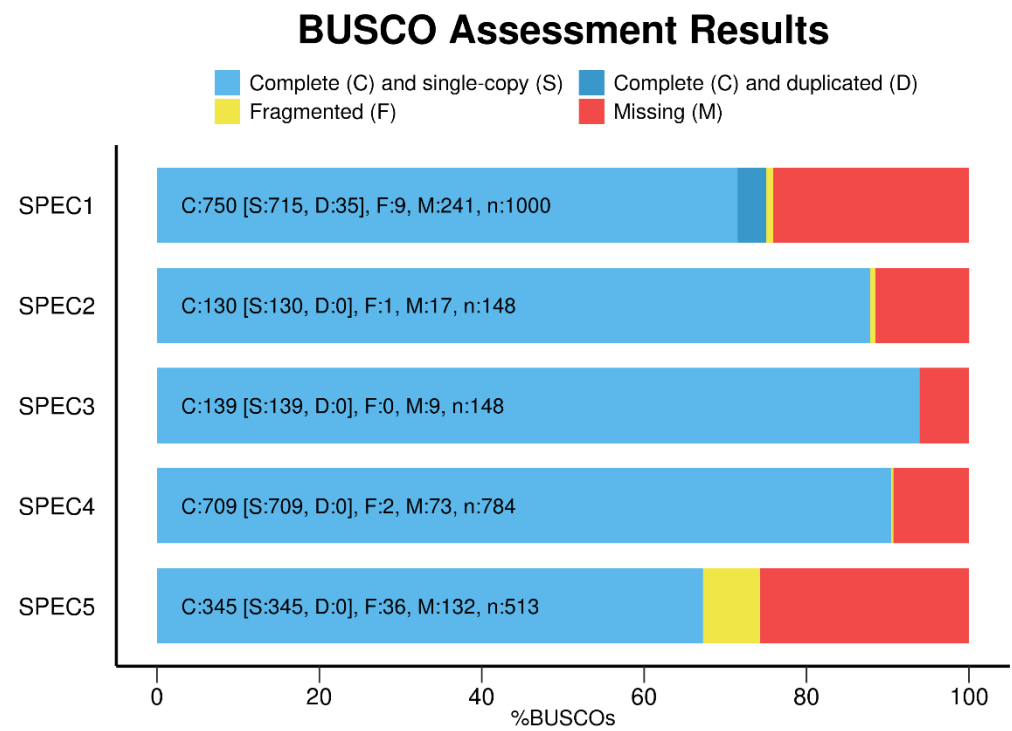
察看结果: 在运行结果文件夹下short_summary_OUTPUT_NAME.txt中有如下统计信息👇1
2
3
4
5
6
7
8C:80.0%[S:80.0%,D:0.0%],F:0.0%,M:20.0%,n:10
8 Complete BUSCOs (C)
8 Complete and single-copy BUSCOs (S)
0 Complete and duplicated BUSCOs (D)
0 Fragmented BUSCOs (F)
2 Missing BUSCOs (M)
10 Total BUSCO groups searched
也可图像化展示结果👇:1
2cp short_summary_OUTPUT_NAME.txt ./plot
python2.7 BUSCO_plot.py -wd ./busco/plot/