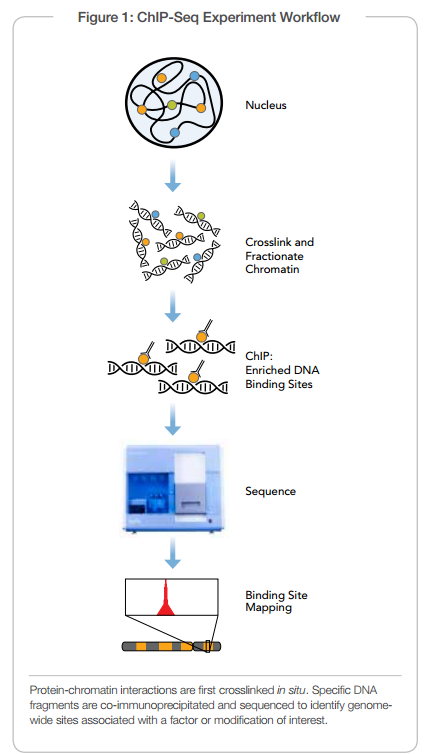
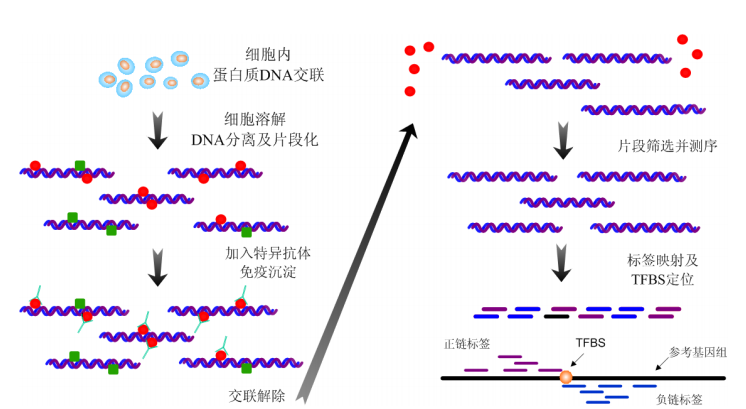
简介
MicroRNAs (miRNAs) are short in sequence and are generated by enzymatic excision from precursor transcripts called primary miRNAs (pri-miRs), which until now had been assumed not to encode any proteins.
Lauressergues et al find that plant pri-miRNAs contain short open reading frame sequences that encode regulatory peptides. This is the first report of a functional peptide being encoded by a pri-miR and provides a fresh perspective on the significance of pri-miR regions beyond those that directly give rise to miRNAs.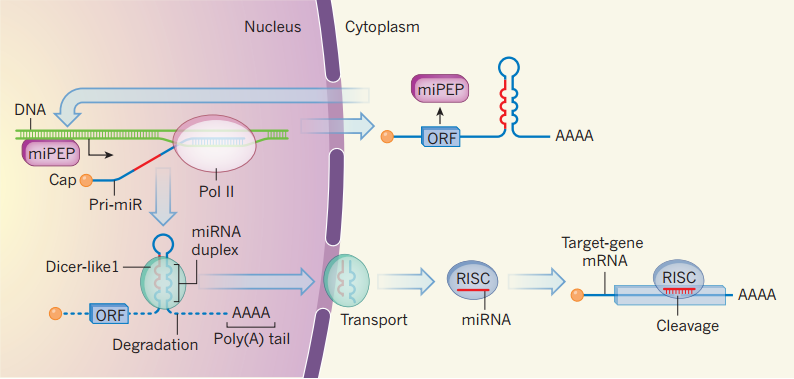
图片注释:MicroRNAs and their associated peptides. The precursors of plant microRNAs (miRNAs) are
pri-miR sequences, which are transcribed from DNA by the enzyme RNA polymerase II (Pol II). They are
then modified by capping and addition of a poly(A) tail. The miRNA duplexes are subsequently excised by
the enzyme Dicer-like1 and transported to the cytoplasm; other parts of the pri-miR are degraded. After
further processing, the resulting miRNA sequence guides repression of gene expression as part of the
RISC complex. Lauressergues et al.report that some pri-miRs contain short open reading frame (ORF)
sequences that can produce peptides (miPEP). The miPEPs enhance expression of the pri-miR, leading
to more miRNA and so more effective cleavage of the target gene’s messenger RNA. Such ORFs may avoid
degradation as part of pri-miRs that might exit the nucleus without being processed by Dicer-like1.
关键信息总结
1) Both plant and animal pri-miRs are transcribed from DNA in the nucleus by the enzyme RNA polymerase II.
2) The structured (fold-back) region of the transcript surrounding the miRNA sequence is recognized and processed by one of two enzymes-Drosha or Dicer-like1(In plants, Dicer-like1 cuts out the miRNA in a duplex form).
3) Transporter proteins export the excised sequences to the cytoplasm, where they are further processed before becoming competent to guide the RNA-induced silencing complex (RISC) in repressing target genes through either cleavage or translational repression of their mRNAs.
4) The miPEPs had the same tissue distribution as their associated mature miRNAs and enhanced the expression and effectiveness of these miRNAs. Moreover, the miPEPs promoted the transcription of their corresponding pri-miR, rather than enhancing miRNA stability.
miRNA合成途径
miRNA的合成大体经过:初级miRNA(pri-miRNA, primary miRNA)→前体miRNA(pre-miRNA, precursor-miRNA)→成熟miRNA(miRNA, mature miRNA)。
其具体合成途径存在两种形式:经典合成途径和Mirtron合成途径;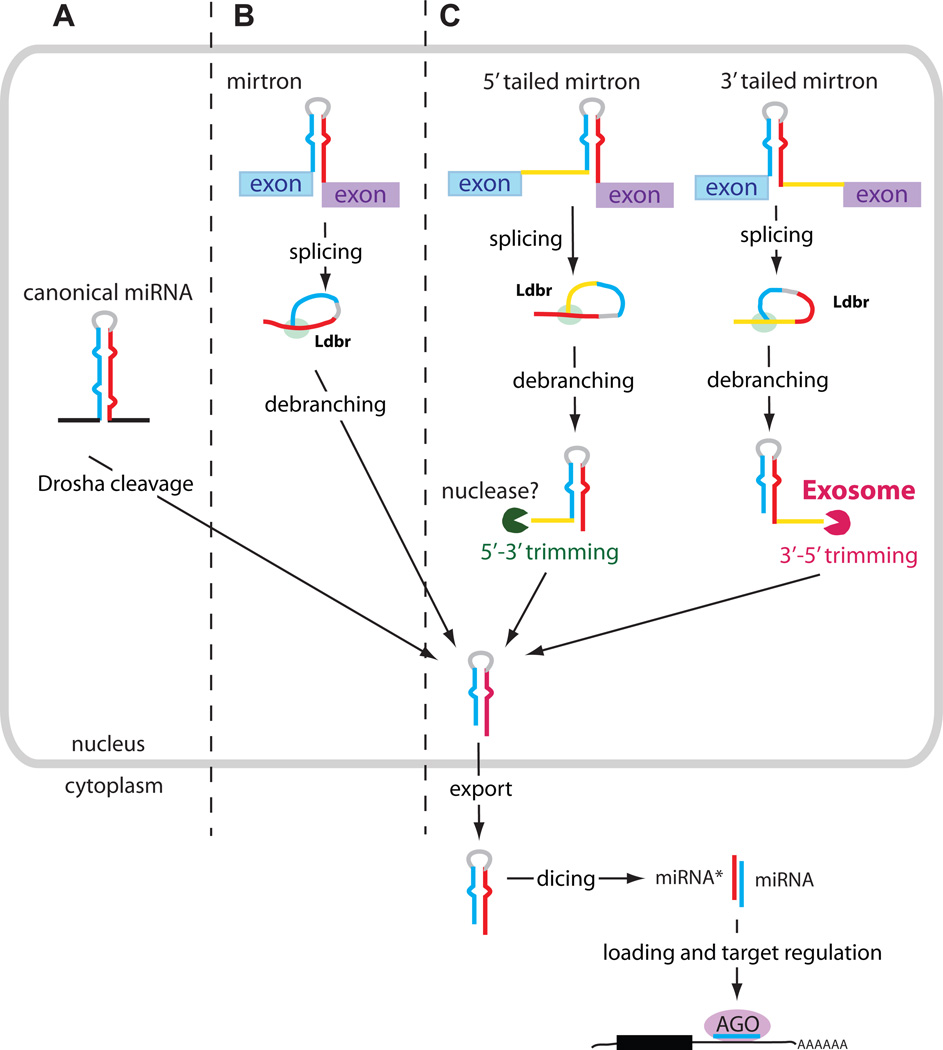
经典合成途径
来源:基因间隔区或编码基因内含子中;
细胞核内编码miRNA 的基因通过RNA聚合酶II 或RNA聚合酶III 转录生成初级miRNA(pri-miRNA),pri-miRNA与来自蛋白质编码基因mRNA相似,有5’ 端帽式结构和3’ 端多聚腺苷酸尾结构,长度可达数千个碱基。接着pri-miRNA在一种RNaseIII(Drosha 酶)和它的伴侣分子(DiGeorge syndrome critical region gene 8,DGCR8)组成的复合物作用下,剪切为70~80 个核苷酸长度、具有茎环结构的miRNA 前体(pre-miRNA)。miRNA 前体在Ran-GTP 依赖的核质/ 细胞质转运蛋白——输出蛋白5(exportin 5)的作用下,从核内运输到胞质中。随后miRNA前体在另一种RNaseIII(Dicer酶)的作用下被剪切成21~25 个核苷酸长度,而且其5’端磷酸化和3’端有2 nt 的悬垂序列,类似于siRNA 的不完全配对的双链RNA,它们是由成熟miRNA与miRNA* 组成的二聚体,miRNA*是pre-miRNA中的一段RNA,其位置恰好与成熟的miRNA 相对。最后在RNA解旋酶作用下生成成熟miRNA 和miRNA*,成熟miRNA结合到RNA诱导的基因沉默复合物(RNA-induced silencing complex, RISC)中发挥作用,miRNA* 则被降解。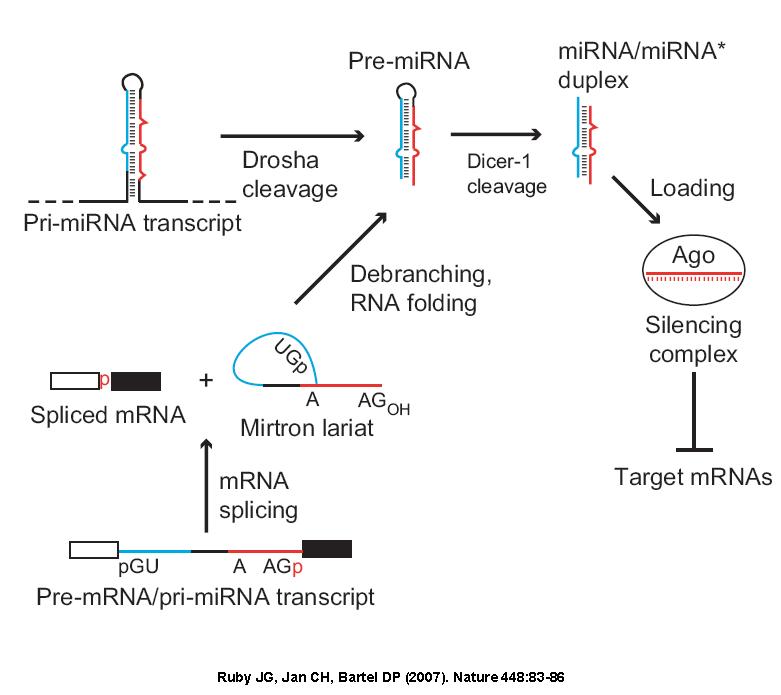
Mirtron途径
2007年,Ruby等在研究黑腹果蝇和秀丽隐杆线虫的小RNA 序列时首次发现mirtron,是一类定位于mRNA编码基因内含子内的miRNA,与经典miRNA合成相比其合成过程不需要经过Drosha酶切割,其形式功能与miRNA相同,具有以下特点:
(1)来自长度为56 nt 的内含子序列;
(2)能够形成miRNA:miRNA*复合物;
(3)内含子序列及其二级结构非常保守;
(4)内含子存在于基因组中的外显子中间并且有”GU-AG”的典型内含子特点;
(5)最后形成的成熟miRNA总来自于3’ 端侧的序列;
(6)合成需要Ldbr(lariat-debranching enzyme,套索分支酶)(参与内含子剪接) 的作用;
(7)与经典的miRNA一样,依赖Ran-GTP/ 输出蛋白5 的转运机制转运出核,在Dicer 酶的作用下成为成熟的miRNA。同时,mirtron在哺乳动物中和在无脊椎动物中有明显的不同,如:
(1)哺乳动物中的mirtron途径合成的功能miRNA大多数来自mirtron的5’茎序列;
(2)不管哺乳动物中的mirtron3’ 茎序列能否成为成熟miRNA,它的起始核苷酸总是尿嘧啶(U)或胞嘧啶(C);
(3)哺乳动物中mirtron悬垂序列(overhang, 发夹结构末端突出序列)大多数为单核苷酸(G),只有少数是双核苷酸(AG);
(4)哺乳动物中mirtron比经典的miRNA前体和无脊椎动物mirtron的GC 含量要高,自由能要低。
阻断miRNA对mRNA的调控作用
miRNA海绵(miRNA sponges)
miRNA海绵是一种miRNA靶基因的竞争性抑制剂,是将若干个miRNA的反义序列串联在一起,连接到合适的载体中表达,其转录物”吸附”相应的miRNA,与miRNA靶基因形成竞争,导致靶基因去抑制化。
miRNA海绵表达载体的构建策略
circRNA
环状RNA(circRNA)对miRNA的吸附作用。http://tiramisutes.github.io/2016/04/12/Circular-RNAs/
miRNA数据库
miocroRNA databases:http://micrornadatabases.com/Databases.html
miRNA相关工具
mirtron prediction
Mirtron’SVM’prediction
MicroRNA Target Prediction
miRanda — miRNA target prediction for human, drosophila and zebrafish genomes
miRBase — a comprehensive repository for miRNAs and their predicted targets
miRDB — an online database for miRNA target prediction and functional annotations in animals
miRNAMap — a genomic maps of microRNA genes and their target genes in mammalian genomes
miR2Disease — a database providing comprehensive resource of miRNA deregulation in various human diseases
TarBase — a comprehensive database of experimentally supported animal microRNA targets
PicTar — microRNA targets for vertebrates, fly and nematodes
TargetScan — a search for the presence of conserved sites that match the seed of each miRNA
Target Gene Prediction at EMBL — miRNA-Target predictions for Drosophila miRNAs
Databases for microRNA Expression
microRNA.org — predicted microRNA targets & target downregulation scores. Experimentally observed expression patterns
HMDD — Human MicroRNA Disease Database (HMDD) is a database that contains the experimentally supported miRNA-disease
association data, which are manually curated from publications. The dysfunction evidence or miRNAs and literature PubMed ID are also given
TransmiR — a web query-driven database integrating the experimentally supported transcription factor and miRNA regulator relations
RNA Secondary Structure Prediction
DIANA MicroTest — a prediction of miRNA-mRNA interaction
mfold — tools for predicting the secondary structure of RNA and DNA, mainly by using thermodynamic methods
microInspector — a web tool for detection of miRNA binding sites in an RNA sequence
miRNA Bioinfor — miRNA End Energy calculator which takes miRNA duplex to calculate free energy for 5 base pairs at
one end plus a dangling nucleotide
miRRim — a method for detecting miRNA foldbacks based on hidden Markov model (HMM)
MXSCARNA — a multiple alignment tool for RNA sequences using progressive alignment based on pairwise structural alignment algorithm of SCARNA. Good for large scale analyses.
RNAhybrid— a tool for finding the minimum free energy hybridisation of a long and a short RNA
MicroRNA Homologous Prediction
miRNAminer — a web-based tool used for homologous miRNA gene search in several species
miRviewer — a global view of homologous miRNA genes in many species
RISCbinder— prediction of guide strand of microRNAs
Mireval — Sequence evaluation of microRNA properties
MicroRNA Deep Sequencing
miRanalyzer— A microRNA detection and analysis tool for next-generation sequencing experiments
miRNAkey— A software pipeline for the analysis of microRNA Deep Sequencing data
miRDeep— Discovering known and novel miRNAs from deep sequencing data
miRNA百科
microRNA.gene-quantification.info
参考文献:
Plant biology: Coding in non-coding RNAs
Mirtrons: microRNA biogenesis via splicing